
Figure 1. Zebrafish larvae. (A) Five-day old (5-day post-fertilization (5-dpf)) zebrafish larva and its prey, a paramecium. (B) 4-dpf nacre larva embedded in 2% low melting-point agarose. Note that the nacre strain lacks black pigments on the body while the retinal pigment epithelium is intact. Scale bar: 0.5 mm. (C) Dissecting needle used for orienting zebrafish larvae in agarose. Scale bar: 1 cm. (D) Screenshot of the software used in the two-photon microscopy, showing "Bleaching", "Time Series", and "Regions" panels that were used in ablation.
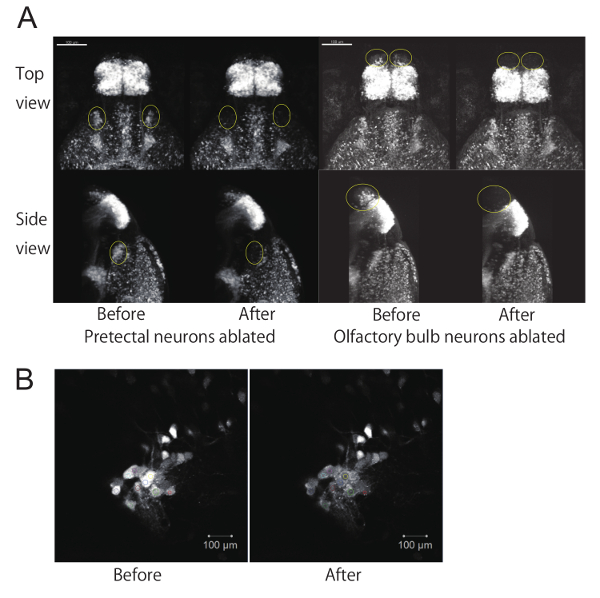
Figure 2. Ablation of a subpopulation of neurons using a two-photon laser microscope. (A) EGFP fluorescent images of 4-dpf zebrafish larvae before and after ablation of neurons. Top view and side view of 3D reconstructed z-stack images using image processing software. The z-stack images were taken with a 20X objective lens. The ablated areas (either the pretectum or olfactory bulb) are circled in yellow. Scale bar: 100 µm. (B) Example of laser ablation at a single focal plane using the "Bleaching" function. Laser-irradiated areas (set as ROIs) are shown in colors on each cell. Scale bar: 10 µm.

Figure 3. Preparation of zebrafish larvae and paramecia for Ca imaging. (A) Recording chamber. A commercially available 9 mm diameter x 0.8 mm depth hybridization gasket with 8 chambers was utilized as the recording chamber. The gasket was put on a glass slide and adhered. The seal on the top of the gasket was peeled off. (B) The nylon mesh (32 µm) used to rinse the paramecia. The mesh was placed on a 50-mL tube. (C) Paramecium culture containing rice straw and dry yeast pellets. (D) Paramecium stock solution prepared from the culture shown in C. (E) Paramecium stock solution was rinsed with system water twice to remove possible olfactory and gustatory cues in the medium. (F) Paramecium embedded in the recording chamber. To remove a portion of agarose, several cuts were made. (G) Recording chamber with a zebrafish larva and a paramecium. The majority of the agarose was removed to allow the paramecium to swim. The head of the zebrafish larva is exposed. (H) Upright fluorescent microscope used in Ca imaging. The microscope is equipped with a scientific CMOS camera. (I) Zebrafish larva in the recording chamber under the microscope with the excitation light on. With a 2.5X objective lens, the illuminated area is slightly larger than the size of the chamber.
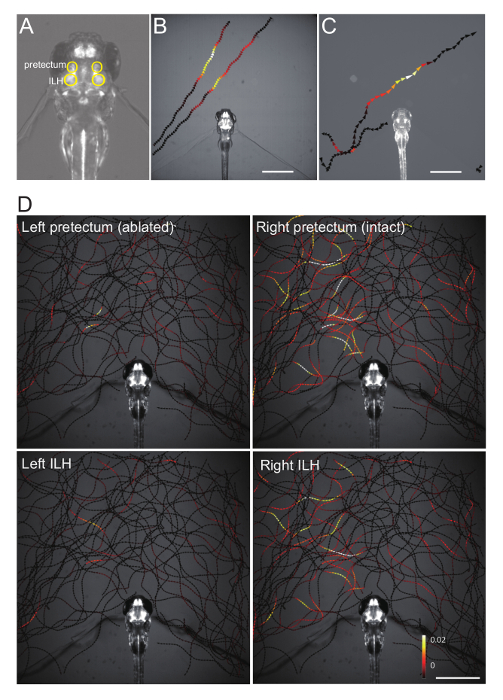
Figure 4. Representative Ca imaging data. (A) Position of the pretectum and the inferior lobe of the hypothalamus (ILH), circled in yellow, in a double Gal4 hspGFFDMC76A; gSAIzGFFM119B; UAShspzGCaMP6s zebrafish larva. (B) Changes in the pretectum Ca signal (averaged bilaterally), color-mapped onto the trajectories of a paramecium in a gSAIzGFFM119B; UAShspzGCaMP6s larva. Scale bar: 1 mm. (C) Changes in the ILH Ca signal (averaged bilaterally), color-mapped on the trajectories of a paramecium in a hspGFFDMC76A; UAShspzGCaMP6s larva. Scale bar: 1 mm. (D) Changes in the pretectum and the ILH Ca signals color-mapped on the trajectories of a paramecium in a hspGFFDMC76A; gSAIzGFFM119B; UAShspzGCaMP6s larva that was subjected to two-photon laser-ablation of the left pretectum. Scale bar: 1 mm. This image was reproduced from the same data previously published.
