Cell Fate Reprogramming of Nematode Germ Cells into Neurons
Abstract
Source: Kolundzic, E., et al. Application of RNAi and Heat-shock-induced Transcription Factor Expression to Reprogram Germ Cells to Neurons in C. elegans. J. Vis. Exp. (2018).
This video demonstrates cell fate reprogramming of nematode germ cells into neurons. Transgenic larvae of the nematode Caenorhabditis elegans are subjected to RNA interference (RNAi) to knock down a specific chromatin-regulating factor, making the germ cells of their progeny more susceptible to cell fate reprogramming. Heat shock is applied to the progeny, inducing the germ cells to express a neuron-specific transcription factor that triggers their transformation into neurons, which is identified using an expressed fluorescent reporter.
Protocol
All procedures involving animal models have been reviewed by the local institutional animal care committee and the JoVE veterinary review board.
1. Solution Preparation
- NGM-Agar plates (1 L)
- Add 3 g of NaCl, 2.5 g of peptone media (e.g., Bacto-Peptone), and 20 g of agar. After autoclaving, add 1 mL of cholesterol (5 mg/mL in 95% ethanol [EtOH] stock), 1 mL of 1 M MgSO4, 1 mL of 1 M CaCl2, 25 mL of 1 M K2PO4, and 1 mL of amphotericin B (2.5 mg/mL stock).
- NGM-Agar RNAi plates (1 L)
- Add 3 g of NaCl, 2.5 g of peptone media, and 20 g of agar. After autoclaving add, 1 mL of cholesterol (5 mg/mL in 95% EtOH stock), 1 mL of 1 M MgSO4, 1 mL of 1 M CaCl2, 25 mL of 1 M K2PO4, 1 mL of amphotericin B (2.5 mg/mL stock), 1 mL of 1 M isopropyl β- d-1-thiogalactopyranoside (IPTG), and 1 mL carbenicillin (50 mg/mL).
- Luria-Bertani (LB)-Agar (1 L)
- Add 10 g of peptone media, 5 g of yeast extract, 5 g of NaCl, 20 g of agar, 10 mL of 1 M Tris pH 8.0. Add H2O to 1 L. After autoclaving, add 1 mL of 50 mg/mL carbenicillin and 2.5 mL of 5 mg/mL tetracycline.
- LB Medium (1 L)
- Add 10 g of peptone media, 5 g of yeast extract, 5 g of NaCl, and 10 mL of 1 M Tris pH 8.0. Add H2O to 1 L. After autoclaving, add 1 mL of 50 mg/mL carbenicillin.
- M9-buffer (1 L)
- Add 6.0 g of Na2HPO4, 3 g of K2PO4, 5 g of NaCl, and 50 mg of gelatin. Before usage, add 1 mL of 1 M MgSO4.
- Bleaching solution (10 mL)
- Add 1 mL of NaClO and 2 mL of 5 N NaOH. Add H2O to 10 mL.
2. Preparation of RNAi Plates
Note: C. elegans is typically cultured in the laboratory on 6 cm Petri plates containing 7.5 mL of Nematode Growth Medium Agar (NGM-Agar). This protocol is optimized for worms kept at 15 °C. To prepare plates with NGM, use standard sterile techniques to prevent fungal and bacterial contamination. The following is the protocol to prepare NGM plates supplemented with reagents for RNAi.
- Prepare 6-cm NGM-Agar RNAi plates containing 1 mM IPTG and 50 µg/mL carbenicillin. Let them dry for 24 – 48 h at room temperature in the dark.
- Keep RNAi plates at 4 °C in the dark. Do not use if older than 14 days.
- Select the RNAi bacteria (Escherichia coli HT115) clone containing the L4440 plasmid with the lin-53 gene DNA sequence from the frozen glycerol stock from the available RNAi library13 on LB-agar plates containing 50 µg/mL carbenicillin and 12.5 µg/mL tetracycline by using the three-phase streaking pattern. Grow the bacteria at 37 °C overnight. This clone allows IPTG-dependent production of lin-53 dsRNA in the bacteria.
- Additionally, grow RNAi bacteria that contain the L4440 plasmid without any gene sequence as the empty vector control.
- The next day, pick a single colony from lin-53 or empty vector LB-agar plates using a 200 µL pipette tip and inoculate each of them into a separate culture tube containing 2 mL of liquid LB medium supplemented with 50 µg/mL carbenicillin. Grow cultures overnight at 37 °C until they reach an optical density (OD) at 600 nm of 0.6 – 0.8. Measure the OD using a spectrophotometer.
CAUTION: The liquid LB media should not contain tetracycline in contrast to the LB-agar plate used for streaking the bacteria from the glycerol stock, since inclusion of tetracycline during feeding leads to a decreased RNAi efficiency. - Add 500 µL of each bacterial culture (lin-53 or empty vector) to 6 cm NGM-Agar RNAi plates using a multipipette. Incubate plates with a closed lid overnight at room temperature in the dark to dry. During this time, the IPTG in the NGM plates will induce production of dsRNA in the bacteria.
- Use at least 3 plates per bacterial culture per experiment. This will provide 3 technical replicates for each experiment.
- Store dried NGM-agar RNAi plates with bacteria at 4 °C in the dark for up to two weeks.
3. Preparation of C. elegans Strain BAT28
- Maintain the worm strain BAT288 containing the transgenes otIs305 [hsp-16.2prom::che-1, rol-6(su1006)] and ntIs1 [gcy-5prom::gfp] on OP50 bacteria using standard NGM-Agar plates at 15 °C.
Note: For detailed protocol on Nematode culture, see. The rol-6(su1006) is a dominant allele and commonly used phenotypic injection marker16 that causes the typical rolling movement. It is used in the otIs305 transgene to track hsp-16.2prom::che-1.- Keep the BAT28 strain at 15 °C at all times in order to prevent precautious activation of the hsp-16.2prom::che-1 transgene. Reduce exposure to temperatures higher than 15 °C while handling the strain as much as possible.
- To age-synchronize worms, use the bleaching technique.
- Wash off 6 cm NGM-Agar plates containing adults and eggs of BAT28 using 900 µL M9 buffer. Pellet worms by centrifuging at 900 x g for 1 min. Remove the supernatant.
- Add 0.5 – 1 mL of bleaching solution and shake the tube for approximately 1 min until adult worms start to burst open. Monitor using a standard stereomicroscope.
- Pellet worms by centrifuging at 900 x g for 1 min. Remove the supernatant.
- Wash the worm pellet 3 times by adding 800 µL of M9 buffer and centrifuging at 900 x g.
- Place the cleaned eggs on fresh NGM-plates seeded with OP50 bacteria. Grow a bleached population on OP50 bacteria at 15 °C until they reach L4 stage (approximately 4 days). L4 larvae can be recognized by a white patch approximately halfway along the ventral side of the worm18 using a standard stereomicroscope.
Note: To achieve the germ cell to neuron conversion phenotype upon depletion of lin-53, it needs to be depleted in the parental generation (P0). The scoring for the conversion phenotype is undertaken in the following generation (filial generation F1). To achieve the deplete lin-53 already in the P0, L4 animals are subjected to RNAi.
- Manually transfer 50 L4 stage worms per replicate using a platinum wire to an NGM plate that does not contain any bacteria and let worms move away from any transferred OP50 bacteria. Let worms move on the plate for around 5 minutes. Use bacteria from the NGM-Agar RNAi plates previously seeded with lin-53 RNAi bacteria (or empty vector control) to transfer to the respective RNAi plates.
- Avoid transferring OP50 bacteria to the actual RNAi plates. Work fast to minimize exposure time of the strain to temperatures higher than 15 °C.
- Incubate worms on NGM-Agar RNAi plates at 15 °C for approximately 7 days until F1 progeny of the worms reaches L3 – L4 stage. They can be clearly separated from the P0 animals since they are bigger and thicker than the F1 progeny.
- Visually check under a standard stereomicroscope whether F1 progeny worms show the protruding vulva (pvul) phenotype as shown in Figure 1. RNAi against lin-53 has pleiotropic effects and causes the pvul phenotype which can be used to assess whether RNAi against lin-53 has been successful.
Note: RNAi against lin-53 can also cause lethality of F1 embryos. This effect increases if plates are exposed to higher degrees than 15 °C before animals reached the L3 – L4 stage. Dead embryos can be recognized by arrested development and lack of hatching. - Incubate plates in the dark since IPTG is light sensitive.
- Visually check under a standard stereomicroscope whether F1 progeny worms show the protruding vulva (pvul) phenotype as shown in Figure 1. RNAi against lin-53 has pleiotropic effects and causes the pvul phenotype which can be used to assess whether RNAi against lin-53 has been successful.
4. Induction of Germ Cell Reprogramming
- Incubate examined RNAi plates containing lin-53 and empty vector RNAi-treated F1 progeny for 30 min at 37 °C in the dark to activate CHE-1. Use a vented incubator since it allows for a more efficient heat-shock.
- Allow heat-shocked animals to recover for 30 min at room temperature in the dark and then incubate plates at 25 °C overnight in the dark.
- Using a standard stereomicroscope coupled to a fluorescence light source, examine animals under the green fluorescent protein (GFP) filter for gcy-5prom::gfp-derived signals in the mid-body area of the worms as shown in Figure 2. Set microscope for GFP signals: excitation maximum at 488 nm with emission maximum at 509 nm.
- Evaluate the degree of germ cell to neuron conversion by mounting animals with GFP signals in the mid-body area on agar pad-containing microscopy slides.
- Count the number of animals showing the germ cell to neuron conversion vs. the dark animals to assess the phenotype penetrance. Typically, around 30% of the animals show discrete GFP signals in the germline upon lin-53 depletion, whereas not more than 5% of the animals show diffuse GFP signals in the germline upon empty vector RNAi.
Representative Results
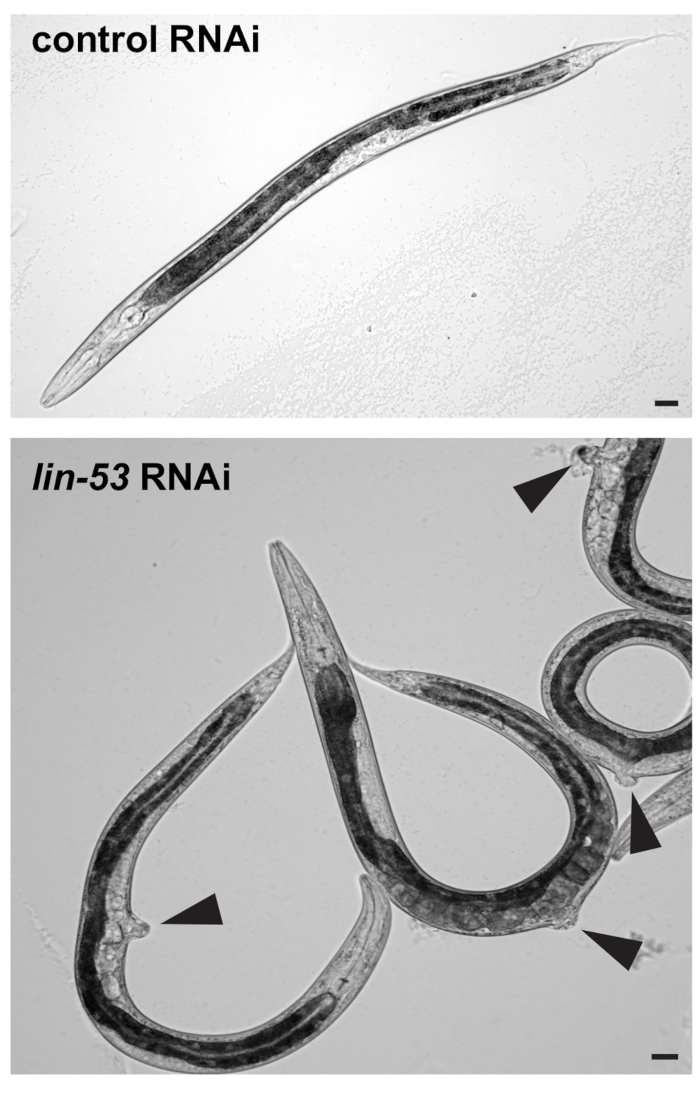
Figure 1: pvul phenotype caused by RNAi against lin-53. (Top) Differential interference contrast (DIC) picture of L4/young adult stage F1 progeny worms derived from control or lin-53 RNAi treated mothers. Scale bars = 20 µm. (Bottom) Animals treated with lin-53 RNAi display the protruding vulva (pvul) phenotype (black arrow-heads). The pvul phenotype confirms that RNAi against lin-53 was successful. Scale bars = 20 µm.
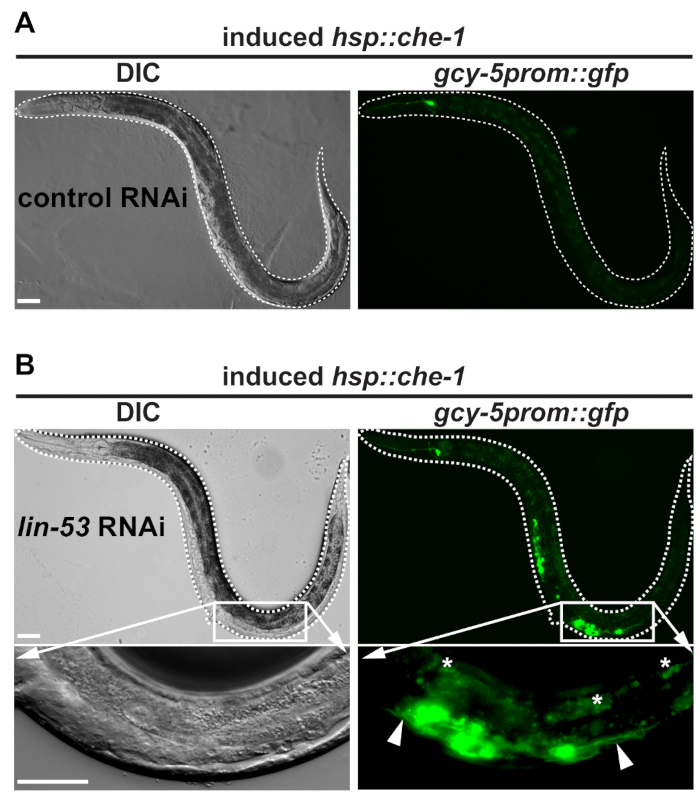
Figure 2: Worms with successfully reprogrammed germ cell into neurons. (A) BAT28 strain animals grown on the empty vector do not show GFP signals in the germline after heat-shock induction of che-1 overexpression. Dashed white line outlines worm. Scale bars = 20 µm. (B) In contrast to BAT28 strain animals grown on the empty vector control RNAi animals grown on lin-53 RNAi display gcy-5prom::gfp signals in the mid-body area. Pictures of the mid-body section with GFP signals taken at 63X magnification reveal GFP-positive cells showing protrusions (white arrow-heads). This morphological feature indicates that germ cells underwent conversion into ASE neuron-like cells. White asterisks mark intestine-derived auto-fluorescence. All pictures were acquired using an epifluorescence microscope with 20X magnification and 63X magnification. Dashed white line outlines worms. Scale bars = 20 µm.
Offenlegungen
The authors have nothing to disclose.
Materials
| Chemicals | |||
| Agar-Agar, Kobe I | Roth | 5210.1 | |
| Bactopeptone | A. Hartenstein GmbH | 211 677 | Peptone Media |
| Yeast extract | AppliChem | A1552,0100 | |
| Amphotericin B | USBiological | A2220 | |
| CaCl2 | Merck Biosciences | 208290 | |
| Cholesterol | Roth | 8866.2 | |
| Gelatine | Roth | 4275.3 | |
| IPTG | Sigma | 15502-10G | |
| K2PO4 | Roth | T875.2 | |
| KH2PO4 | Roth | 3904.2 | |
| MgSO4 | VWR | 2,51,63,364 | |
| Na2HPO4 | Roth | P030.1 | |
| NaCl | Roth | 9265.1 | |
| NaClO | Roth | 9062.3 | |
| NaOH (5 N) | Roth | KK71.1 | |
| Tris | Roth | AE15.2 | |
| Carbenicillin | Roth | 634412 | |
| Tetracycline | Roth | 2371.2 | |
| Incubators | |||
| Incubator | New Brunswick Scientific C24 | M1247-0052 | for heat-shock |
| Incubator | Sanyo MIR-5 | 5534210 | for maintenance of worm strains at 15 °C or 25 °C |
| Microscopes | |||
| Fluorescence microscope | M205 FA | Leica | |
| Microscope (Imaging) + camera | Axio Imager 2 | Zeiss | |
| Microscope (Imaging) + camera | Sensicam | PCO | |
| Stereomicroscope | SMZ745 | Nikon | |
| Bacterial strains | |||
| Escherichia coli HT115 | F-, mcrA, mcrB, IN(rrnD-rrnE)1, rnc14::Tn10(DE3 lysogen: lavUV5 promoter -T7 polymerase) (IPTG-inducible T7 polymerase) (RNAse III minus). | ||
| Escherichia coli OP50 | Uracil auxotroph E. coli strain | ||
| Worm strains | |||
| BAT28: otIs305 (hsp16.2prom::che-1::3xHA) ntIs1 (gcy-5prom::gfp) V. | derived from OH9846 by 4x times more back crossing with N2 | ||
| RNAi Clones | |||
| lin-53 | SourceBioscience | ID I-4D14 | Ahringer library 16B07 ChromI, K07A1.12 |
| empty vetor: L4440 | Addgene | #1654 | |
| Misc. | |||
| Worm pick | self-made using a pasteur pipette and a platinum wire |
