מקור:ג’ונתןפ. בלייז 1 , אליזבת סוטר1, וכריסטופר פ קורבו1
1 המחלקה למדעי הביולוגיה, מכללת וגנר, דרך הקמפוס 1, סטטן איילנד ניו יורק, 10301
הערכה כמותית של פרוקריוטים יכולה להיות מכבידה בהתחשב בשפע שלהם, נטייתם להתפשטות מעריכית, מגוון מינים באוכלוסייה וצרכים פיזיולוגיים ספציפיים. בנוסף לאתגר זה, הוא הטבע הארבע-פאזי שבו חיידקים משתכפלים (עיכוב, יומן, נייח ומוות). היכולת להעריך במדויק את ריכוז המיקרואורגניזם נחוצה לזיהוי מוצלח, בידוד, טיפוח ואפיון (6). ככזה, מיקרוביולוגים השתמשו בדילול סדרתי וטכניקות ציפוי שונות במשך למעלה ממאה שנה כדי לכמת באופן אמין עומס חיידקי ויראלי בסביבות מעבדה קליניות, תעשייתיות, פרמצבטיות ואקדמאיות (2,4,6). תיאורים של מתודולוגיה זו הופיעו לראשונה בשנת 1883 כאשר המדען והרופא הגרמני רוברט קוך פרסם את עבודתו על סוכנים הגורמים למחלות זיהומיות (2). הטכניקות הידועות מראש של קוך, המכונות לעתים קרובות אבי הבקטריולוגיה המודרנית, הפכו לסטנדרט הזהב לספירת מיקרואורגניזמים, הניתנים לפולחן או אחר, ברחבי העולם.
דילול סדרתי הוא הפחתה שיטתית של ישות ידועה או לא ידועה (solute, אורגניזם, וכו ‘) באמצעות השעיה מחדש רצופה של פתרון ראשוני (פתרון0) לתוך נפחים קבועים של דילול נוזלי (ריק). החסרים האלה מורכבים בדרך כלל 0.45% מלוחים, אם כי הרכב יכול להיות מגוון (7). בעוד שנסיין יכול לבחור כל אמצעי אחסון עבור כל דילול, זה לרוב כפולה של 10, להקל על הפחתה לוגריתמית של המדגם. לדוגמה, פתרון0 מכיל סך של 100 תאי E. coli מושעים ב 10 מ”ל של מרק מיזיני. אם 1 מ”ל של פתרון0 מוסר ומתווסף 9 מ”ל של מלוחים (דילול1),הפתרון החדש (פתרון1)יכיל 1/10th של הריכוז הראשוני של E. coli. בדוגמה זו, הפתרון החדש (פתרון1) יכיל 10 תאי E. coli. חזרה על תהליך זה על ידי הסרת 1 מ”ל של פתרון1 והוספתו עוד 9 מ”ל של מלוחים (דילול2) יניב פתרון2, המכיל רק תא E. coli יחיד. מכיוון שכל פתרון חדש (9 מ”ל של דילול + 1 מ”ל של פתרון) מכיל סך של 10 מ”ל, אנו יכולים להסיק כי גורם הדילול להפחתה זו הוא 10 או שזה היה דילול סדרתי פי 10 (איור 1). מכיוון שהתחלנו רק עם 100 תאים בדוגמה זו ואנחנו מדללים בגורם של 10, רק שני שלבים נדרשים כדי להגיע לריכוז המינימלי המוחלט של תא אחד.
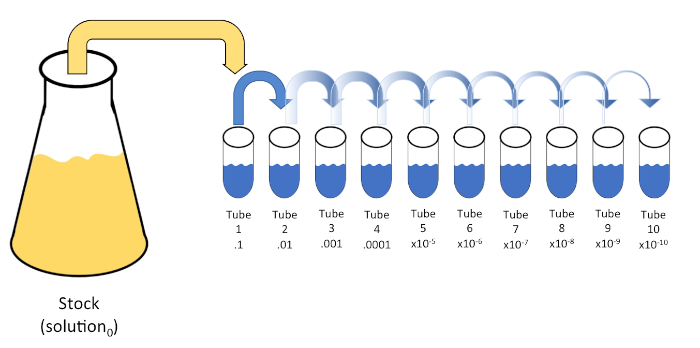
איור 1: דילול סדרתי של פתרון מניות. 1 מ”ל aliquot של פתרון המניה (פתרון0) מתווסף צינור 1 אשר מכיל 9 מ”ל של 0.45% מלוחים (dilent1); המוצר של תערובת זו הוא פתרון1. חזור על ידי aliquoting 1 מ”ל של פתרון חדש שנוצר1 והוספתו צינור 2. Aliquoting ו resuspension ממשיך בצורה זו עד הצינור הסופי הוא הגיע, דילול ריכוז המניה על ידי גורם של 10 כל אחד עם כל צעד. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
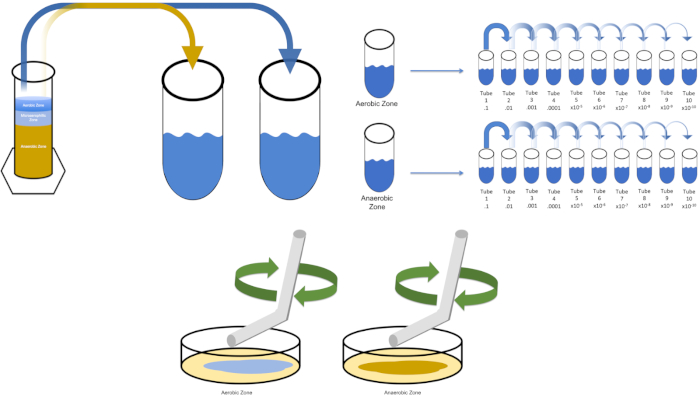
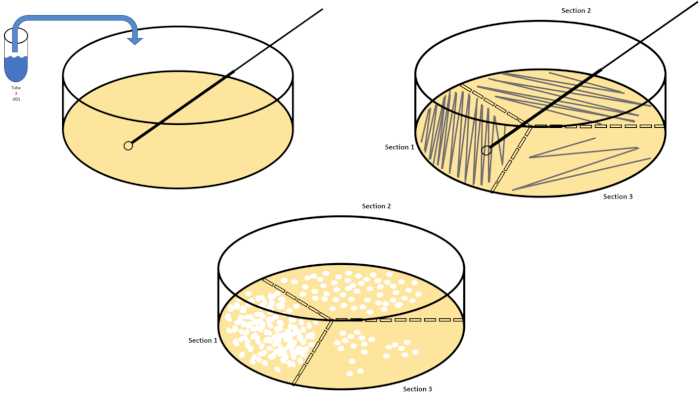
דילול סדרתי הוא הטכניקה הפשוטה ביותר להשגת ריכוזים לניהול של אורגניזם רצוי והוא משלים פסים של צלחת פטרי והתפשטות, רק שתיים מטכניקות ציפוי רבות המשמשות מיקרוביולוגים. יתרון זה של גישה זו הוא כי הנסיין יכול לקצור זנים טהורים של מין אחד או זנים נפרדים מאוכלוסייה מעורבת (7). פסים מתבצעים על ידי החדרת אורגניזם למדיום מוצק (בדרך כלל מורכב אגרוז) כי הוא יגדל על אם החומרים המזינים המתאימים זמינים. ניקוי עדין של לולאת חנק סטרילית על פני המדיום (כך שיישאר פס עדין) בתבנית סינוסואידית נוקשה, תפיץ את האורגניזם באופן פרופורציונלי לתדירות צורת הגל של הנסיין. חלוקת צלחת הפטרי לשליש או לרבעים (רצף רבעים) והפחתת התדירות של כל פס כאזור חדש של המנה נכנס בהדרגה להפחית את מספר מיקרואורגניזמים שיכולים לכבוש את האזור הזה, לייצר מושבות בודדות במקום דשא חיידקי בלתי ניתן לערעור. ציפוי ממרח אינו מדלל בנוסף דגימות; מפיץ זכוכית סטרילי משמש להפצת עליקוט של מדיית השעיה על פני צלחת פטרי שלמה(איור 2). המושבות הגדלות על צלחת ההתפשטות נובעות מתא יחיד וניתן לספור כל מושבה בצלחת כדי להעריך את מספר היחידות היוצרות מושבה למיליליטר (CFU) בהשעיה נתונה, המיוצגת כ- CFU / mL (6) (איור 3) אגר רך וציפוי משוכפל הם וריאציות של הטכניקות הנ”ל ומאפשרים בידוד של בקטריופאז ‘והקרנה מוטנטית, בהתאמה (1,7).
איור 2: צלחת פסים לספירת חיידקים ובידוד מתחים. סמן את תחתית צלחת הפטרי עם פרטי זיהוי (שם חיידק, תאריך, מדיה) וחלק לשליש. לאחר בחירת דילול מתאים של מדגם המלאי, לקחת לולאת חנק סטרילית (חד פעמית או להבה) ולהטביע אותו במבחנה (כאן, T3). להעלות מעט את כיסוי צלחת פטרי בצד אחד, כך רק לולאה מכחיק יכול לגשת אגר. גלה את הלולאה המחוסנת על פני החלק העליון של התקשורת בצורה זיג-זג נזהר לא להתפשר על אגר. סובב את הלוח בערך פי1/3 (~ 118°) והפחית את תדירות תנועת זיג-זג. סובב בפעם האחרונה והפחת את תדירות הזיג-זג פעם נוספת. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
איור 3: למרוח ציפוי. גרם אחד של האזור האירובי היה תוחם מחדש ב- T1 ולאחר מכן מדולל באופן סדרתי. זכוכית סטרילית או מוט מתפשט חד פעמי פלסטיק משמש להפצת אינוקולום בכל מנה. זה חזר על עצמו עם 1 גרם של האזור אנאירובי. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
כמו בדילול סדרתי, קנה מידה לוגריתמי משמש כדי לבטא ריכוז אורגניזמי. ניתן למנות ידנית את מספר המושבות הגדלות בצלחות פטרי סטנדרטיות בגודל 100 מ”מ x15 מ”מ (או אוטומטיות בעזרת עיבוד חישובי) על ידי זיהוי אשכולות מבודדים של צמיחה. ספירות הכוללות פחות מ- 30 או יותר מ- 300 צריכות להיות מוגדרות כמעטות מכדי לספור (TFTC) או רבות מכדי לספור (TNTC), בהתאמה. במקרה של האחרון, דילול סדרתי צריך להתבצע כדי להפחית את הריכוז לפני restreaking צלחת פטרי חדשה. ממוצע מספר המושבות העצמאות שזוהו משלוש מנות פטרי נפרדות והכפלת הממוצע על ידי גורם הדילול יניבו CFU / mL; התווייתיומן 10 של CFU / mL נגד הזמן תחשוף את זמן הדור הממוצע של האורגניזם (7).