连续稀释和电镀:微生物枚举
English
分享
概述
资料来源:乔纳森·布莱泽1号,伊丽莎白·苏特1号,克里斯托弗·科博1号
1大学生物科学系,瓦格纳学院,1 校园路,纽约州斯塔顿岛,10301
鉴于原核生物的丰度、指数增殖倾向、种群内的物种多样性以及特定的生理需求,对原核生物的定量评估可能十分繁重。使这一挑战更加复杂的是细菌复制的四相性质(滞后、原木、静止和死亡)。准确估计微生物浓度的能力对于成功识别、隔离、培养和表征(6)是必要的。因此,微生物学家在一个多世纪以来一直采用连续稀释和各种电镀技术,在临床、工业、制药和学术实验室环境中可靠地量化细菌和病毒载量(2,4,6)。1883年,德国科学家和医生罗伯特·科赫(Robert Koch)发表了他关于致病剂(2)的著作。科赫的引种技术通常被称为现代细菌学之父,已成为全世界可培养或其他微生物的祭标标准。
连续稀释是通过连续将初始溶液(溶液0)重新悬浮成液体稀释剂(空白)的固定体积,系统地还原已知或未知的实体(溶质、有机体等)。这些空白通常由0.45%的盐水组成,虽然成分可以变化(7)。虽然实验者可以为每个稀释剂选择任意体积,但它通常是 10 的倍数,有助于对数减少样本。例如,溶液0共含有100个大肠杆菌细胞,悬浮在10 mL的营养汤中。如果去除溶液0的1mL并加入9 mL的盐碱(稀释剂1),新溶液(溶液1)将含有大肠杆菌初始浓度的1/10。在此示例中,新溶液 (溶液1) 将包含 10个大肠杆菌细胞。重复这个过程,通过去除1mL的溶液1,并将其添加到另外9 mL的盐水(稀释剂2)将产生溶液2,只包含一个大肠杆菌细胞。由于每种新溶液(9 mL稀释剂 = 1 mL溶液)总共含有10mL,我们可以得出结论,这种还原的稀释系数为10倍,或者这是10倍的序列稀释(图1)。由于我们在本例中只从 100 个细胞开始,并且我们稀释了 10 倍,因此只需两个步骤即可达到 1 个细胞的绝对最小浓度。
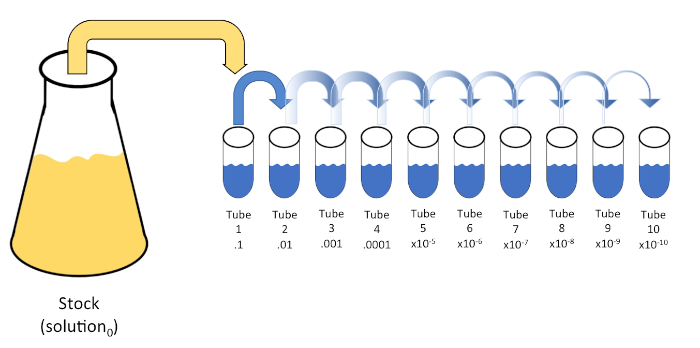
图1:库存溶液的串行稀释。在管1中加入1 mL等分的库存溶液(溶液0),其中含有9 mL的0.45%盐碱(盐分1);这种混合物的产物是溶液1。重复此操作,将新创建的解决方案1的 1 mL 等分,并将其添加到管 2 中。同位和再悬浮继续这种方式,直到达到最终管,稀释股票浓度每一步10倍。请点击此处查看此图的较大版本。
连续稀释是获得所需生物体的可管理浓度的最简单技术,辅以培养皿条纹和扩散,只是微生物学家使用的许多电镀技术中的两种。这种方法的好处是,实验者可以收获单个物种的纯菌株或与混合种群分离菌株(7)。条纹是通过将一个有机体引入固体介质(通常由甘蔗素组成)来完成的,如果适当的营养物质可用,它会生长在这种介质上。以刚性正弦模式轻轻扫过介质的无菌接种回路(使细微的条纹保持),将生物体与实验者波形的频率成比例分布。将培养皿分成三分之三或四分之一(象限条纹),并在进入菜的新区域时降低每个条纹的频率,将逐渐减少可以占据该区域的微生物数量,产生单一菌落,而不是不可量化的细菌草坪。铺板不额外稀释样品;无菌玻璃扩张器用于在整个培养皿中分配悬架介质的等分(图2)。生长在扩散板上的菌落来自单个细胞,盘上的每个菌落可以计算以估计给定悬浮液中每毫升(CFU)的菌落形成单位数,表示为CFU/mL (6) (图3)软琼脂和复制品电镀是上述技术的变化,允许分离噬菌体和突变筛选,分别(1,7)。
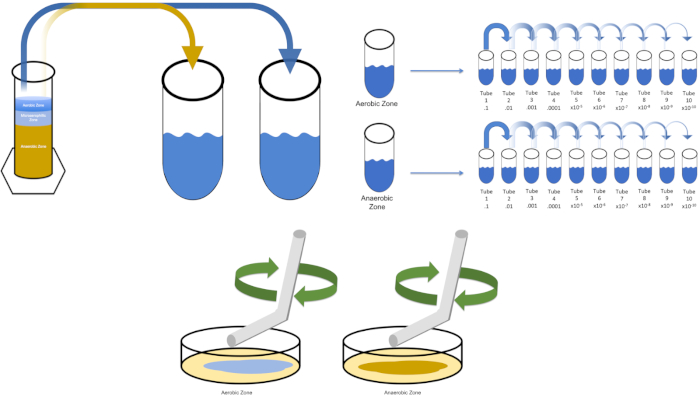
图2:用于细菌枚举和菌株分离的板条纹。用识别信息(细菌名称、日期、介质)标记培养皿的底部,然后分成三份。选择适当的稀释库存样品后,采取无菌(一次性或火焰)接种回路并将其浸入试管(此处,T3)。稍微提高一侧的培养皿盖,以便只有接种回路才能进入琼脂。以锯齿形的方式将接种回路滑过介质顶部,小心不要损害琼脂。将板旋转大约 1/3rd (±118°),并降低锯齿形运动的频率。旋转最后一次,并再次降低锯齿频率。请点击此处查看此图的较大版本。
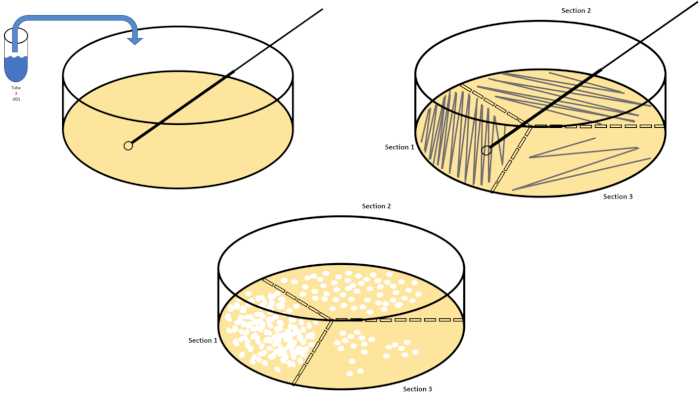
图3:铺板。1克有氧区在T1中重新悬浮,然后连续稀释。无菌玻璃或塑料一次性摊杆用于在每个菜盘中分配接种液。这与1克的厌氧区重复。请点击此处查看此图的较大版本。
与连续稀释一样,使用对数尺度来表达有机体浓度。在标准培养皿中生长的菌落数量为100mm x15mm,可通过识别孤立的生长簇进行手动枚举(或借助计算处理实现自动化)。总数少于 30 或大于 300 的计数应分别定义为计数太少(TFTC) 或计数数量过多而无法计数 (TNTC)。在后者的情况下,应进行连续稀释,以降低浓度,然后再重新处理新的培养皿。从三个单独的培养皿中识别的自足菌落的数量平均乘以稀释系数的平均值将产生CFU/mL;绘制CFU/mL的日志10与时间将揭示生物体的平均生成时间(7)。
Procedure
Applications and Summary
Bacterial enumeration and strain isolation by plating requires manageable concentrations of target organisms. Successful plating is therefore contingent upon serial dilution. As such, the aforementioned techniques remain the cornerstone of microbiological examination and experimentation. Though simple by design, dilution factors and plating technique can be modified to by the experimenter to bolster outcomes without compromising the integrity of each method. Plotting the four phases of bacterial growth can be helpful when characterizing desired microbes. These phases, lag, log, stationary, and death, are marked by changes in bacterial replication. The lag phase features slow growth due to physiological adaptation, the log phase is the period of maximum proliferation featuring an exponential rise in viable cells, stationary phase is then reached due to environmental limitations and accumulations of toxins, before the death phase where cell counts begin to fall. This can be accomplished by serially diluting (or 1-step diluting to avoid confusion) Solution0 every hour for a total of 8 hours, beginning at Time0 (Solution0 should be returned to a shaking incubator after each dilution). Calculate the log10 of CFU/ml for a single diluent of Time0 and plot on the Y-axis. Repeat this calculation for the sample Time1 (make sure calculate CFU/mL using the same dilution factor as Time0). Repeat until each time (Time1-Time8) are plotted on the X-axis.
References
- Allen, M.E., Gyure, R.A. (2013) An Undergraduate Laboratory Activity Demonstrating Bacteriophage Specificity. Journal of Microbial Biological Education 14: 84-92.
- Ben-David, A., Davidson, C.E. (2014) Estimation Method for Serial Dilution Experiments. Journal of Microbiological Methods 107:214-221.
- Goldman, E., Green, L.H. (2008) Practical Handbook of Microbiology.
- Koch, R. (1883) New Research Methods for Detection of Microcosms in Soil, Air and Water.
- Lederberg, J., Lederberg, E.M. (1952) Replica Plating and Indirect Selection of Bacterial Mutants. Journal of Bacteriology 63:399-406
- Pepper, I., Gerba, C., Ikner, L. (2019) Bacterial Growth Curve Analysis and its Environmental Changes. JoVE Science Education Database. Environmental Microbiology.
- Sanders., E.R. (2012) Aseptic Laboratory Technique: Plating Methods. JoVE 63:e3063.
成績單
Sometimes, in order to identify and study bacteria we first need to isolate and enrich them from a sample. For example, samples obtained from a Winogradsky Column are mixed, meaning they contain multiple species or strains of bacteria, so studying an individual bacterium or enumerating the different kinds present can be challenging. To this end, serial dilution and plating techniques are typically employed to reliably quantify bacterial load and isolate individual colonies.
Serial dilution is a process through which the concentration of an organism, bacteria in this example, is systematically reduced through successive resuspension in fixed volumes of liquid diluent. Usually the volume of the diluent is a multiple of 10 to facilitate logarithmic reduction of the sample organism. For example, one gram of sediment is first removed from the Winogradsky zone of interest and added to 10 milliliters of an appropriate liquid medium. Then, one milliliter of this first dilution is added to another tube containing nine milliliters of medium. The process can be repeated until several different concentrations of bacteria have been prepared. Serial dilution is the key to enumeration of bacteria in this example, since mixed samples from a Winogradsky Column contain an unknown, often large, number of bacteria.
Next, streak plating and spread plating enable the isolation and enumeration of bacteria within a sample, respectively. Streaking is accomplished by introducing a diluted sample to one section of the solid medium supplemented with nutrient, which is divided into thirds. This inoculum is then spread over each third of the plate in a zig-zag pattern. As different sections of the plate are streaked, crossing from the previous sample only once, the sample is spread more thinly. This means that you may only need to streak from one dilution to achieve individual colonies in the later sections. After incubation, the streaked plates allow for observations of colony morphology, information that can help differentiate between different bacterial species.
Alternatively, if the main goal is the enumeration of the bacteria in the sample spread plating may be used. In spread plating, an aliquot of a single sample is spread evenly over the entire surface of solid medium. Typically, because we don’t know the bacterial numbers in the mixed sample, a spread plate is made for each of the dilutions or a representative sample of them. After incubation, enumeration can be performed using these spread plates. Any plates with colony counts fewer than 30 should be discarded, since small counts are subject to greater error. Similarly, any counts over 300 should be discarded because colony crowding and overlapping can lead to underestimation of colony count. If the colony counts of each of these remaining dishes is recorded and multiplied by the dilution factor, and then divided by the volume plated, this yields the colony forming units, or CFUs, per milliliter of suspension. In this video, you will learn how to qualitatively and quantitatively evaluate a sample containing a known bacterium, and the microbial communities contained in various regions of a Winogradsky Column via serial dilution, spread plating, and streak plating.
First, put on any appropriate personal protective equipment including a lab coat, gloves, and goggles. Next, sterilize the workspace with 70% ethanol and wipe down the surface. Next, gather two 500 milliliter Erlenmeyer flasks and label one broth and the other agar. To prepare LB agar solution, mix approximately 6.25 grams of LB agar, three grams of technical agar, and 250 milliliters of distilled water in the flask labeled agar.
Then, prepare LB broth by combining 2. 5 grams of LB media and 100 milliliters of distilled water in the flask labeled broth. After autoclaving the flasks, use a heat resistant glove to remove the flasks from the autoclave and place them in a 40 to 50 degree Celsius water bath. Once the flasks are 50 degrees Celsius, carefully prepare three 100 milliliter aliquots of the broth solution and label each aliquot solution zero. Next, gather 10 sterile petri dishes and label them with the date, name, type of media used, and the Winogradsky Column zone that the organisms will be harvested from. Pipette 15 milliliters of agar from the agar flask into each petri dish. Then, use the pipette tip to remove any bubbles, replace the plate lids, and allow them to solidify on the bench top overnight.
The next day, wipe down the bench top with 70% ethanol. Next, label 10 20 milliliter test tubes T1 through T10 and place them in a rack. Pipette nine milliliters of .45% saline into each tube. Now, cover each of the 10 test tubes loosely with their caps and transfer them to an autoclave-compatible test tube rack. After the cycle is complete, remove the saline blanks using heat resistant gloves and allow them to cool. Store the tubes at room temperature until they have reached approximately 22 degrees Celsius.
To cultivate a known target organism, E. coli in this example, inoculate 100 milliliters of solution zero with a single colony from a previously streaked plate. Then, cover the tube and incubate it over night at 37 degrees Celsius. To evaluate the regions of a Winogradsky Column, add approximately one gram of material from the aerobic zone to T1 and resuspend by vortexing. Then, repeat this process with one gram of material from the anaerobic zone.
Remove the tube containing solution zero inoculated with E. coli from the incubator and shake it. Then, pipette one milliliter of the solution into a T1 test tube and vortex to mix. Remove one milliliter of solution from T1 and transfer it to T2, vortexing to mix. Repeat this process through tube T10. To evaluate the aerobic and anaerobic zones of the Winogradsky Column, remove one milliliter of solution from each of the previously prepared T1 tubes and transfer it to the appropriate T2 tubes. Then, continue the serial dilutions through the T10 tubes as previously demonstrated.
To spread plate, pipette 100 microliters of the diluted sample from each T3 tube on to the corresponding petri dish. Then, use a sterile spreading rod to gently distribute the sample on to the petri dish and replace the plate lid. Repeat this process for the T6 and T9 dilutions, as previously demonstrated. Incubate the plates containing aerobic organisms in a 37 degree Celsius incubator for 24 hours. Incubate the plates containing anaerobic organisms in an anaerobic chamber set to 37 degrees Celsius for 24 hours. The next day remove the T3, T6, and T9 dilution plates from the incubator and the anaerobic chamber and transfer them to the bench top. Working with one plate at a time, glide a sterile inoculating loop across the top of the media in a zig-zag pattern. Then, replace the petri dish lid. Next, rotate the plate by 1/3 and sterilize the loop to reduce the frequency of the previously made zig-zag pattern. Again, after sterilizing the loop, rotate the plate by 1/3, reduce the frequency of the zig-zag pattern one last time, and replace the lid. Repeat this streaking method for the remaining plates, as previously shown. Then, place the streaked plates containing aerobic organisms in a 37 degree Celsius incubator overnight and the streaked plates containing anaerobic organisms in an anaerobic chamber set to 37 degrees Celsius overnight.
Cultures were harvested from the aerobic and anaerobic zones of a seven day Winogradsky Column. Then, the cultures were serially diluted prior to streaking and spreading on LB agar plates. Streaking revealed a mixed population from each of the evaluated Winogradsky zones, and the spread plates produced similar results. A plate streaked from a mixed population will result in bacterial colonies of different shapes, sizes, textures, and colors. In contrast, the streaked and spread plates containing the known organism, E. coli, demonstrated a homologous population. Generally, it is best to calculate CFUs per milliliter using the average colony count of three plates spread with the same sample and dilution factor. Multiply the average number of colonies by the dilution factor and divide by the amount aliquoted. Finally, isolated colonies chosen from each plate can be used in further enrichment assays to determine species identity.