A Cell Culture Model for Studying the Role of Neuron-Glia Interactions in Ischemia
Summary
Here, we present a simple approach using specific culture media that allows the establishment of neuron- and astrocyte-enriched cultures, or neuron-glia cultures from the embryonic cortex, with high yield and reproducibility.
Abstract
Ischemic stroke is a clinical condition characterized by hypoperfusion of brain tissue, leading to oxygen and glucose deprivation, and the consequent neuronal loss. Numerous evidence suggests that the interaction between glial and neuronal cells exert beneficial effects after an ischemic event. Therefore, to explore potential protective mechanisms, it is important to develop models that allow studying neuron-glia interactions in an ischemic environment. Herein we present a simple approach to isolate astrocytes and neurons from the rat embryonic cortex, and that by using specific culture media, allows the establishment of neuron- or astrocyte-enriched cultures or neuron-glia cultures with high yield and reproducibility.
To study the crosstalk between astrocytes and neurons, we propose an approach based on a co-culture system in which neurons cultured in coverslips are maintained in contact with a monolayer of astrocytes plated in multiwell plates. The two cultures are maintained apart by small paraffin spheres. This approach allows the independent manipulation and the application of specific treatments to each cell type, which represents an advantage in many studies.
To simulate what occurs during an ischemic stroke, the cultures are subjected to an oxygen and glucose deprivation protocol. This protocol represents a useful tool to study the role of neuron-glia interactions in ischemic stroke.
Introduction
According to data from the World Health Organization, about 5.5 million people die every year from ischemic stroke1. This condition is characterized by the interruption of blood flow to a certain brain region, resulting in a reversible or irreversible loss in the supply of oxygen and nutrients to the tissue, which alters tissue function and leads to mitochondrial dysfunction, calcium dysregulation, glutamate excitotoxicity, inflammation and cell loss2,3.
Apart from vascular cells, neuronal and glial cells are involved in the pathophysiology of the ischemic stroke4. In particular, astrocytes are essential to the maintenance of neurons and recently were shown to play a critical role in the response to the ischemic lesion5. This type of glial cell performs functions of structural support, defence against oxidative stress, synthesis of neurotransmitters, stabilization of cell-cell communication, among others6. Along with neurons, astrocytes play a direct role in synaptic transmission, regulating the release of molecules such as adenosine triphosphate, gamma-aminobutyric acid and glutamate7. Part of the injury induced by ischemia is caused by the excessive release of glutamate and its accumulation at the synaptic cleft, leading to the overactivation of N-methyl-D-aspartate receptors, activating downstream signalling cascades, ultimately resulting in excitotoxicity8. Since astrocytes are able to remove glutamate from the synaptic cleft and convert it into glutamine, they are crucial in defending against excitotoxicity, thereby exerting a neuroprotective effect on ischemia. These cells also play a role in ischemia-induced neuroinflammation. After the ischemic insult, activated astrocytes undergo morphologic changes (hypertrophy), proliferate, and show an increase in glial fibrillary acidic protein (GFAP) expression. They can become reactive (astrogliosis), releasing pro-inflammatory cytokines such as tumour necrosis factor-α, interleukin-1α and interleukin-1β, and producing free radicals, including nitric oxide and superoxide, which in turn can induce neuronal death9,10. In contrast, reactive astrocytes may also play a neuroprotective effect, since they release anti-inflammatory cytokines, such as transforming growth factor-β, that is upregulated after stroke11. Moreover, they can generate a glial scar, which can limit tissue regeneration by inhibiting axonal sprouting; however, this glial scar can isolate the injury site from viable tissue, thus preventing a cascading wave of uncontrolled tissue damage12,13.
Thus, it is imperative to establish models that allow studying neuron-glia interactions under an ischemic injury in order to find therapeutic strategies that limit or reverse the effects of ischemic injury. Compared to other models used to study ischemic injury, namely in vivo models14,15,16, organotypic cultures17,18,19 and acute brain slices20,21,22, primary cell cultures are less complex, which makes possible the study of individual contributions of each cell type in the pathophysiology of ischemic stroke and how each cell type responds to possible therapeutic targets. Typically, in order to study the interactions between neuron-enriched cultures and astrocyte-enriched cultures, neurons and glial cells of postnatal origin are used23,24, or postnatal glial cells and embryonic neurons25,26. Herein is proposed a simple approach to establish neuron- or astrocyte-enriched cultures and neuron-glia cultures from the same tissue. These primary cells are obtained from rat embryonic cortex, a region frequently affected by stroke27,28. Moreover, the dissociation of the tissue is performed only by a mechanical procedure. Therefore, this protocol allows isolating cells in the same stage of development, in a fast and inexpensive way, and with high performance and reproducibility.
The crosstalk between astrocytes and neurons can be explored using a co-culture system in which neurons cultured in coverslips are maintained in contact with a monolayer of astrocytes seeded in multiwell plates. Small paraffin spheres can be used to ensure the separation of the two cell cultures. This approach allows independent manipulation of each cell type before they are brought into contact. For example, it is possible to silence a specific gene in astrocytes and see how it can influence the neuronal vulnerability or protection against ischemic-induced damage. An established method to induce ischemic-like conditions in vitro is oxygen and glucose deprivation (OGD)3, which consists in replacing the regular atmosphere (95% air and 5% CO2) by an anoxic atmosphere (95% N2 and 5% CO2), associated with the omission of glucose.
The method described is suitable for studying the interactions between neurons and astrocytes in the context of ischemic stroke, in a simple, fast, reproducible and inexpensive way.
Protocol
All animals used were bred at the CICS-UBI Health Science Research Centre in accordance with the national ethical requirements for animal research and with the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes (Directive 2010/63/EU).
1. Rat embryo cortex primary cell culture
- Culture medium preparation
- Prepare the Neurobasal Medium (NBM) by adding the following supplements: 2% B27, 0.5 mM glutamine, 25 µM glutamate and 120 µg/mL gentamicin. Homogenize, adjust the pH to 7.2 and sterilize the medium with a vacuum filtration step, using a 0.22 µm filter. For the neuron-glia cell culture, supplement the NBM cell culture medium with 10% of Heat-Inactivated Fetal Bovine Serum (HI-FBS).
- Prepare the Minimum Essential Medium Eagle (MEM) medium with the following supplements: sodium hydrogen carbonate 2.2 g/L, insulin from bovine pancreas 5 mg/L, D-glucose anhydrous 3.4 g/L, penicillin (12 U/mL) /streptomycin (12 µg/mL) and 10% HI-FBS. Homogenize, adjust the pH to 7.2 and sterilize the medium with a filtration step.
- Preparation of materials and equipment
- Puncture the terminal part of a 1 mL plastic micropipette tip from side to side, using a needle with a specific diameter (0.5 mm for S; 0.6 mm for M; 0.8 mm for L) and seal the opening of the tip using a flame.
- Sterilize all the glassware. Throughout the dissection keep the tools used (e.g., scissors, tweezers, scalpel) immersed in 70% ethanol. Spray the materials with 70% ethanol before entering them in the laminar flow chamber.
- Set the water bath temperature to 37 °C and place the cell culture medium in the water bath before starting the procedure.
NOTE: For immunocytochemistry assays, poly-D-lysine coating should be performed in multiwell plates containing coverslips.
- Rat embryonic cortex culture
- Remove the rat embryos from a female Wistar rat with 15-16 days of gestation (the end of the mating, which should last 24 h, is considered the 1st day of the embryonic development). For that purpose, anaesthetize the female with ketamine (87.5 mg/kg) and xylazine (12 mg/kg) and remove the embryos. Then euthanize the female rat by cervical dislocation, following standard protocol.
- Place the embryos in a 50 mL sterile tube, add phosphate buffer saline (PBS) until it covers the embryos and quickly take them to the culture room.
- Still inside the yolk sac, place the embryos in a Petri dish containing 25 mL of cold PBS. With the help of scissors and tweezers, break the yolk sac, remove the embryo and transfer it to another Petri dish containing also cold PBS. The PBS in the Petri dish should be enough to cover the entire embryo.
NOTE: Be careful when opening the yolk sac to avoid damaging the embryo. In 1.3.3 and 1.3.4 we used 90 mm diameter Petri dishes placed on top of ice packs covered with absorbent paper to keep the PBS at low temperature. - For dissection of the embryo, transfer it to another Petri dish containing 30 mL of cold PBS. Place the embryo under a dissecting microscope and immobilize it using a tweezer. Make the initial incision parallel to the cortex, going from the ocular cavity to the end of the muzzle and be careful not to decapitate the animal.
- Carefully remove the scalp and the meninges using tweezers, in order not to damage the cortical brain tissue. Make the next incision to separate the cortex. Transfer the cortical tissue to a 15 mL tube containing 5 mL of PBS using a Pasteur pipette.
- Perform the mechanical digestion of the cortical brain tissue using the 1 mL plastic tips prepared in 1.2.1. Triturate 10 times with a regular pipette and repeat the process using pipettes with progressively smaller holes (L, M and S), until the chunks have fallen apart.
- After the mechanical digestion, centrifuge the suspension at 400 x g for 3 min. Discard the supernatant and resuspend the sediment with the appropriate cell culture medium previously warmed at 37 °C.
- Determine the total number of cells present in the cell suspension (cell density) using a Neubauer chamber, make the appropriate dilutions and plate the cells. The initial cell density was defined based on a previous study5.
- For a neuron-enriched culture, use 0.21 x 106 cells/cm2 as the initial cell density and maintain the cells in NBM culture medium without HI-FBS.
- For the neuron-glia cultures use 0.14 x 106 cells/cm2 as the initial cell density and maintain the cells in NBM culture medium supplemented with 10% HI-FBS.
- For an astrocyte-enriched culture, use 0.26 x 106 cells/cm2 as the initial cell density and maintain the cells in MEM supplemented as previously indicated.
- Place the cells in an incubator set at 37 °C, 95% O2 and 5% CO2.
NOTE: For longer culture periods, an anti-mitotic such as 27 μM 5-fluoro-2′-deoxyuridine with 68 μM uridine should be added to suppress cell growth.
2. Co-culture system
- Preparation of materials
- Heat the paraffin at 150 °C in a heating block for approximately 7 min. Keep at 150 °C until finishing the procedure. Then, with the help of a 1 mm diameter sterile glass Pasteur pipette, add a small drop over sterilized and PDL-coated coverslips. The paraffin spheres are irregular, but they have approximately 2 mm diameter. The spheres will allow the two cultures to be separated by approximately 1.25 mm.
- As the paraffin is not sterile, place the multiwell with the paraffin spheres under ultraviolet radiation for 15 min.
- To establish the co-culture, transfer the coverslip with the paraffin spheres using a tweezer previously immersed in 70% ethanol for 15 min.
- Co-culture
- When the two cultures are ready to use (i.e., after 7 days in culture under the conditions mentioned in step 1.3.8), transfer the neurons seeded in the coverslips with paraffin spheres to the wells containing the astrocytes.
- 24 h before the two cultures are brought in contact, change the culture medium of neurons and astrocytes to NBM supplemented, or not, with HI-FBS, depending on the purpose of the experiment.
- After placing both cell types in contact, wait 8-12 h before starting the different stimuli and procedures.
NOTE: The schematic representation of the co-culture system is shown in Figure 1.
3. Oxygen and glucose deprivation
- Culture Medium Preparation
- For the OGD experiments, use Hank's Balanced Salt Solution (HBSS). Prepare the HBSS medium with the following reagents: 1.26 mM CaCl2, 5.36 mM KCl, 0.44 mM KH2PO4, 0.49 mM MgCl2, 139.9 mM NaCl, 4.17 mM NaHCO3, 3.38 mM Na2HPO4. Homogenize and adjust the pH to 7.2. Sterilize the medium by filtration.
NOTE: If appropriate, supplement the HBSS solution with glucose (HBSSglu+), by adding 5.56 mM glucose. For cultures submitted to OGD do not supplement the HBSS medium with glucose (HBSSglu-).
- For the OGD experiments, use Hank's Balanced Salt Solution (HBSS). Prepare the HBSS medium with the following reagents: 1.26 mM CaCl2, 5.36 mM KCl, 0.44 mM KH2PO4, 0.49 mM MgCl2, 139.9 mM NaCl, 4.17 mM NaHCO3, 3.38 mM Na2HPO4. Homogenize and adjust the pH to 7.2. Sterilize the medium by filtration.
- OGD procedure
- 7 days after seeding the cells, remove the culture medium and wash two times with HBSSglu-. After washing add the HBSSglu- cell culture medium and place the multiwell in the hypoxia chamber.
- Seal the hypoxia chamber and add a gas mix containing 95% N2/5% CO2 for 4 min with a flow of 20 L/min to remove the oxygen present inside the chamber. After this, stop the flow and place the hypoxia chamber in an incubator at 37 °C for 4 h or 6 h, depending on the extent of the intended ischemia.
- After the period of OGD, replace the HBSSglu- medium with the appropriate culture medium for the remaining procedures.
NOTE: The OGD experiment aims to simulate the in vitro conditions that the cells suffer during an ischemic event, so it is important to certify that all the media used previously is removed.
4. Immunocytochemistry assay
NOTE: Perform the immunocytochemistry assay as previously described5.
- Briefly, to characterize the different cortical cultures, incubate the cells overnight at 4 °C with rabbit anti-GFAP (1:2000) and mouse anti-microtubule-associated protein 2 (MAP2; 1:500); and then 1 h at room temperature with the following secondary antibodies: anti-rabbit conjugated to Alexa Fluor 546 and anti-mouse conjugated to Alexa Fluor 488, both at 1:1000 dilution.
- Label the cell nuclei by incubation with 2 µM Hoechst 33342 for 10 min at room temperature.
- Mount coverslips in fluorescence mounting medium and acquire images on an epifluorescence microscope with a 63x magnification.
5. Statistical analysis
- Express data as percentage of the total number of cells or as a percentage of control and presented as the mean ± standard error of the mean (SEM) of at least 3 independent experiments performed in triplicate.
- Perform statistical analysis with software (GraphPad Software Inc., San Diego, CA), using the unpaired Student’s t test. The results were considered significant when values of p < 0.05.
Representative Results
To characterize the cultures, immunocytochemistry to assess the number of cells that expressed GFAP or MAP2, widely used markers of astrocytes and neurons (Figure 2), was performed in each type of cortical culture. This analysis revealed that astrocyte-enriched cultures presented 97% of the cells expressing GFAP (Figure 2A). Regarding the neuron-enriched culture 78% of the cells expressed MAP2, 4% of the cells expressed GFAP, and 18% of the cells were both GFAP and MAP2-negative (Figure 2B). In relation to the neuron-glia cortical culture, 49% of the cells were MAP2-positive, 31% were GFAP-positive and 20% were negative for both markers (Figure 2C).
Seven days after establishing the cortical culture, the neuron-glia culture and the neuron-enriched culture were subjected to the OGD procedure, for 4 h or 6 h. After this procedure, the number of MAP2 and GFAP-positive cells was assessed by immunocytochemistry. In the neuron-glia culture, the loss of MAP2-positive cells was 30% and 60% after 4 h and 6 h of OGD, respectively (Figure 3B), while the loss of GFAP-positive cells was 9% and 17% after 4 h and 6 h of OGD, respectively (Figure 3C). Regarding the neuron-enriched culture, there was a decrease of 41% and 64% in the number of MAP2-positive cells after 4 h and 6 h of OGD, respectively (Figure 3A). Moreover, in the neuron-enriched culture, there was a slight increase in the injury extension induced by 4 h of OGD when compared to the neuron-glia culture (Figure 3A).
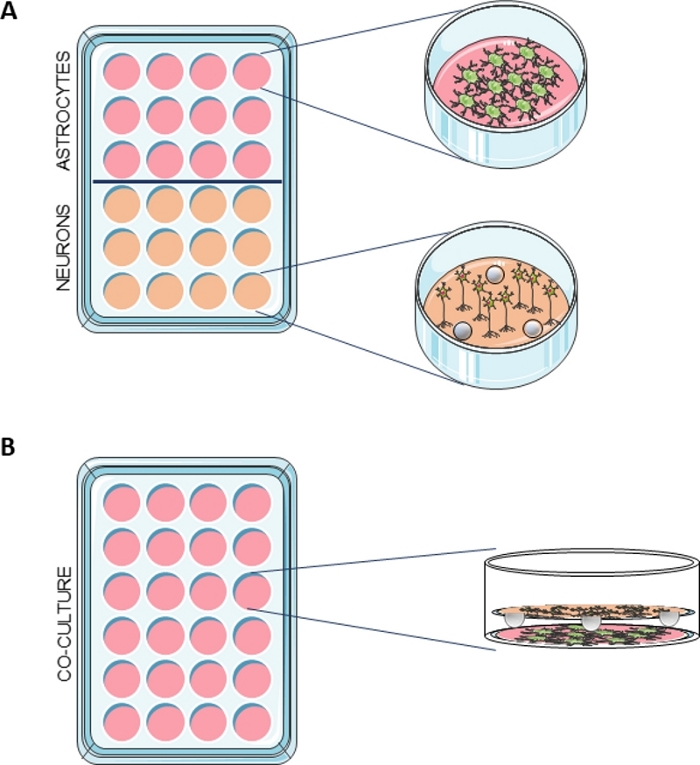
Figure 1: Schematic representation of the co-culture system.
(A) Astrocytes were seeded in a multiwell containing PDL-coated coverslips and neurons were seeded in a multiwell with PDL-coated coverslips containing 3 paraffin spheres. (B) When the two cultures were ready to use, the neurons in the coverslips with the spheres were transferred to the wells containing the astrocytes. Please click here to view a larger version of this figure.

Figure 2: Characterization of neuron-glia cortical culture, neuron-enriched cortical culture and astrocyte-enriched cortical culture.
Percentage of neurons (MAP2-positive cells), percentage of astrocytes (GFAP-positive cells), and percentage of double-negative cells (MAP2-negative/GFAP-negative cells) at the 7th day in culture in (A) astrocyte-enriched culture (B) neuron-enriched culture and (C) neuron-glia culture and representative images showing the immunostaining for MAP2 (green) and GFAP (red). The total number of cells was assessed by quantifying Hoechst 33342-labelled nuclei with non- pyknotic morphology (blue). Due to the low number of neurons in the astrocyte-enriched cortical culture, the representative image does not show MAP2-positive staining. The data are presented as mean ± SEM of 3 independent experiments (A) and 6 independent experiments (B, C) performed in triplicate. Images were acquired with a 63x objective. Please click here to view a larger version of this figure.
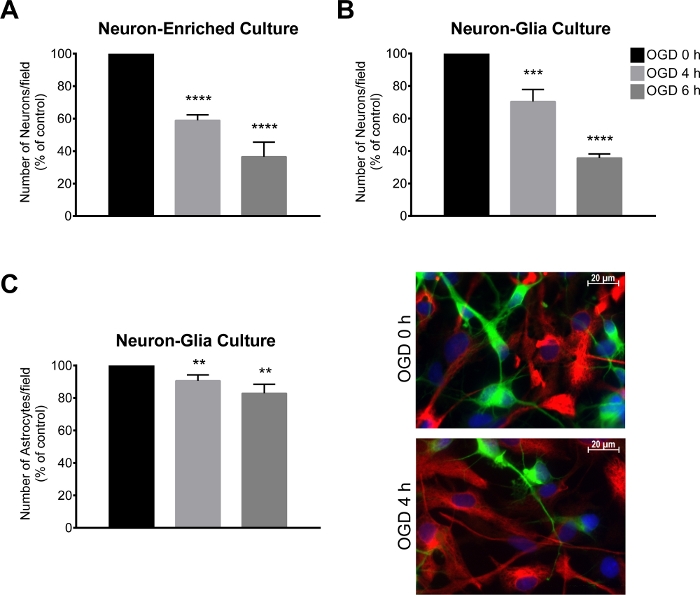
Figure 3: Assessment of neuronal loss following an OGD period.
(A, B) Number of neurons/field (MAP2-positive cells) in a neuron-glia culture and neuron-enriched culture and (C) number astrocytes/field (GFAP-positive cells) and representative image of MAP2 (green) and GFAP (red) immunostaining in a neuron-glia culture. The total number of cells was assessed by quantifying Hoechst 33342-labelled nuclei with non- pyknotic morphology (blue). The neuron-glia and neuron-enriched cultures were submitted to oxygen and glucose deprivation (OGD) for a period of 4 h and 6 h. The data are presented as mean ± SEM of at least 3 independent experiments performed in triplicate. The total number of cells was assessed by quantifying Hoechst 33342-labelled nuclei. **p < 0.01, ***p < 0.001 and ****p < 0.0001 compared to OGD 0 h (unpaired Student’s t test) Please click here to view a larger version of this figure.
Discussion
The method here described consists of the astrocyte and neuron isolation from rat embryonic cortical tissue, allowing the establishment of neuron- or astrocyte-enriched cultures or neuron-glia cultures. It was adapted from a previous study of our group5, where the cortical neuron–glia and neuron-enriched embryonic cultures isolation were described and the two cultures characterized. Using these cultures, Roque et al. found that astrocytes play a key role in responding to an ischemic damage and suggests that communication between astrocytes and neurons is essential to neuroprotection5. In the present protocol, in addition to the preparation of neuron–glia and neuron-enriched cultures, we are also able to obtain astrocytes-enriched cultures, which allows us to study the effect of an ischemic environment on the neurons and astrocytes isolated or together.
Analysis of the immunocytochemistry data showed that 18% of the cells in the neuron-enriched culture and 20% in neuron-glia cultures were negative for both MAP2 and GFAP. These cells presented nuclei with non-pyknotic morphology. Given that the cultures were prepared from embryonic tissue, part of the cells may not yet express the neuronal marker, needing further maturation. This is in line with previous studies indicating that MAP2 expression increases with neuronal maturity and that the number of MAP2-positive cells increased with the time in culture and with the age of the embryos at the time of dissection29,30. We have previously demonstrated that in neuron-glia culture only 0.7% of cells were positive for the microglial marker ionized calcium-binding adapter molecule 15. Although the culture medium used in neuron-glia cultures has the nutritional support necessary for glial cell growth, the amount of microglia in the cortex of embryos with 15-16 days is reduced and as the culture time is reduced, the growth of this cell type is limited. The same applies to neuron-enriched cultures, but in this case the growth of glial cells is even more limited due to the absence of HI-FBS in the culture medium.
In addition to allowing different types of cultures to be obtained from the same preparation, the protocol here described has other advantages. The single-cell suspension is obtained simply by mechanical digestion, unlike other methods that use both enzymatic and mechanical digestion24,25,31,32; therefore, it is faster and cheaper. Another advantage is that this protocol can also be used to prepare cells from other brain regions, such as the hippocampus or the midbrain, allowing the study of pathologies affecting different areas of the brain. Moreover, the alternative procedure described, that allows the establishment of co-cultures, allows the analysis of biochemical and morphological changes that occur in specific cell types present in the co-culture by using methods such as immunocytochemistry. A common model for the establishment of co-cultures is transwell systems24,25,33,34. Contrary to what occurs with a co-culture system using small spacers, such as the paraffin spheres, transwell co-culture models do not allow to perform immunocytochemistry on both cells types present in the co-culture. In addition, the co-cultures using spacers such as the paraffin spheres are simple and low cost.
Subjecting neuron-enriched or neuron-glia cultures to OGD is a common in vitro model for ischemia, nonetheless other in vitro methods have been used, namely chemical and enzymatic methods or induction of excitotoxicity by glutamate3,35. Compared to other methods, OGD allows the simulation of the two phases that occur during the ischemic stroke, namely the deprivation of oxygen and glucose and the reperfusion, which is an advantage because it mimics what occurs in vivo. Moreover, although chemical and enzymatic methods may be useful due to its quick response and ease of application, there is a concern with the relevance to the in vivo pathological state, because chemical hypoxia leads to more free radical generation than anoxia, surpassing what is observed in vivo35. Regarding the OGD protocol, we observed that it leads to neuronal loss and that the extension of the lesion can be adjusted by altering the duration of the OGD period, in order to reach the experimental requirements. The differences observed in neuronal loss after the OGD period in neuron-enriched cultures and neuron-glia cultures might be due to the protective role played by astrocytes, thereby attenuating the neuronal death.
As expected, OGD damage to astrocytes in neuron-glia cultures was lower at both 4 h and 6 h when compared to neurons. The higher resistance of astrocytes to OGD is attributed to multiple aspects. They are able to maintain ATP levels longer than neurons during ischemia, and severe ionic dysregulation proceeds more slowly36: firstly because neurons have higher density of ionic channels and a consequent greater energy demand to maintain ionic gradients; and secondly because most of the glycogen stores in the brain is found in astrocytes36. Additionally, astrocytes express lower levels of ionotropic glutamate receptors than neurons and have better ionic buffering and antioxidant capacity36. These attributes presumably underlie the well-known selective loss of neurons over astrocytes36.
Concerning the limitations of the protocol proposed here, the most significant is that it is based on an in vitro model that lacks the complexity of the interactions that occur in an in vivo system, which can cause translatability issues to an in vivo situation. However, it presents the advantages associated with cell cultures, namely simplicity, ease of manipulation, the capability to provide basic detailed information about how a specific cell population responds to a certain insult3. In vitro models of disease are less time consuming and less expensive to maintain than in vivo models. More specifically, for the modelling of the ischemic stroke, an in vitro model also possesses the advantage of being easier to control the glucose and oxygen levels when compared with the in vivo alternatives34. Furthermore, we also propose the use of co-cultures, which can give a higher level of complexity, allowing to study the interaction between different cell types present in a tissue.
There are some critical steps that require further attention when executing the protocol. Due to the nutritional requirement of astrocytes, the NBM used for obtaining neuron-glia cultures should be supplemented with 10% of HI-FBS-containing growth factors, amino acids and fatty acids. This supplementation is what differentiates the neuron-glia culture from the neuron-enriched culture. To prepare an astrocyte-enriched culture a medium devoid of supplements required for neuronal growth, such as B27, should be used. In the current protocol, the medium of election for the astrocyte-enriched culture was MEM. It is also very important that the needs of the different cell types are ensured when they are brought into contact. For this purpose, a culture medium compatible with both astrocytes and neurons, namely the NBM supplemented with B27 and HI-FBS, may be used. Regarding the OGD protocol, the main critical steps are the removal of all the O2 from the chamber before starting the OGD period, and the proper washing of the cells with HBSS without glucose, in order to eliminate all the glucose present in the medium.
In conclusion, here we present an in vitro model to study the ischemic stroke established in a simple, fast, inexpensive and reproducible way. Additionally, the method described also allows to implement neuron- and astrocyte-enriched primary cultures but also neuron-glia cultures, thus providing a great in vitro model for modelling several brain diseases, with a higher level of complexity than immortalized cell lines and pure neuronal or glial cultures.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors acknowledge the funding support by Fundação para a Ciência e a Tecnologia through Projects UIDB/00709/2020, POCI-01-0145-FEDER-029311 and the fellowship SFRH/BD/135936/2018 to JP, by ‘‘Programa Operacional do Centro, Centro 2020” through the project CENTRO-01-0145-FEDER-000013 and funding to the PPBI-Portuguese Platform of BioImaging through the Project POCI-01-0145-FEDER-022122.
Materials
| 24 -well culture plates | Thermo Fischer Scientific | 142475 | |
| 95% N2/5% CO2 gas cylinder | ArLíquido | ||
| Anti-mouse conjugated to Alexa Fluor 488 | Invitrogen | A11001 | 1/1000 dilution; incubation period – 1 h at room temperature |
| Anti-rabbit conjugated to Alexa Fluor 546 | Invitrogen | A11010 | 1/1000 dilution; incubation period – 1 h at room temperature |
| B27 supplement (50x) | Gibco | 17504-044 | |
| Dako Fluorescence Mounting Medium | Dako | S3023 | |
| D-glucose anhydrous | Fisher Scientific | G/0450/60 | 3.4 g/L |
| Epifluorescence microscope | Zeiss | AxioObserver Z1x | 63x objective |
| Fetal Bovine Serum (FBS) | Biochrom | S0615 | 10% |
| Gentamicin | Sigma-Aldrich | G1272 | 120 µg/mL |
| Glutamate | Sigma-Aldrich | G8415 | 25µM |
| Glutamine | Sigma-Aldrich | G3126 | 0.5 mM |
| Hoechst 33342 | Invitrogen | H1399 | 2 µM; incubation period – 10 min at room temperature |
| Hypoxia incubation chamber | Stemcell Technologies | 27310 | Chamber used for OGD induction |
| Insulin from bovine pancreas | Sigma-Aldrich | I5500 | 5 mg/L |
| Ketamine | Sigma-Aldrich | K-002 | 87.5 mg/Kg |
| Minimum Essential Medium Eagle medium | Sigma-Aldrich | M0268 | warm up to 37 °C before use |
| Mouse Anti-MAP2 | Santa Cruz Biotechnology | Sc-74421 | 1/500 dilution; incubation period overnight at 4 °C |
| Neurobasal medium | Gibco | 21103-049 | warm up to 37 °C before use |
| Paraffin pastilles for histology | Sigma-Aldrich | 1.07164 | Solidification point 56-58°C |
| Paraformaldehyde | Sigma -Aldrich | P6148 | 4% in PBS |
| Penicilin/Streptomycin | Biochrom | A 2213 | penicillin (12U/mL) /streptomycin (12µg/mL) |
| Poly-D-lysine | Sigma-Aldrich | P1024 | |
| Rabbit Anti-GFAP | DAKO | Z0334 | 1/2000 dilution; incubation period overnight at 4 °C |
| Sodium hydrogen carbonate | Fisher Scientific | S/4240/60 | 2.2g/L |
| Xylazine | Sigma-Aldrich | X1126 | 12 mg/Kg |
References
- Donkor, E. S. Stroke in the 21(st) Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Research and Treatment. 2018, 3238165 (2018).
- Dirnagl, U., Iadecola, C., Moskowitz, M. A. Pathobiology of ischaemic stroke: an integrated view. Trends in Neurosciences. 22 (9), 391-397 (1999).
- Woodruff, T. M., et al. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Molecular Neurodegeneration. 6 (1), 11 (2011).
- Berezowski, V., Fukuda, A. M., Cecchelli, R., Badaut, J. Endothelial cells and astrocytes: a concerto en duo in ischemic pathophysiology. International Journal of Cell Biology. 2012, 176287 (2012).
- Roque, C., Baltazar, G. Impact of Astrocytes on the Injury Induced by In vitro Ischemia. Cellular and Molecular Neurobiology. 37 (8), 1521-1528 (2017).
- Ransom, B. R., Ransom, C. B. Astrocytes: multitalented stars of the central nervous system. Methods in Molecular Biology. 814, 3-7 (2012).
- Halassa, M. M., Fellin, T., Haydon, P. G. The tripartite synapse: roles for gliotransmission in health and disease. Trends in Molecular Medicine. 13 (2), 54-63 (2007).
- Forder, J. P., Tymianski, M. Postsynaptic mechanisms of excitotoxicity: Involvement of postsynaptic density proteins, radicals, and oxidant molecules. 神经科学. 158 (1), 293-300 (2009).
- Basic Kes, V., Simundic, A. M., Nikolac, N., Topic, E., Demarin, V. Pro-inflammatory and anti-inflammatory cytokines in acute ischemic stroke and their relation to early neurological deficit and stroke outcome. Clinical Biochemistry. 41 (16-17), 1330-1334 (2008).
- Gursoy-Ozdemir, Y., Can, A., Dalkara, T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke. 35 (6), 1449-1453 (2004).
- Cekanaviciute, E., et al. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia. 62 (8), 1227-1240 (2014).
- Huang, L., et al. Glial scar formation occurs in the human brain after ischemic stroke. International Journal of Medical Sciences. 11 (4), 344-348 (2014).
- Rodriguez-Grande, B., et al. The acute-phase protein PTX3 is an essential mediator of glial scar formation and resolution of brain edema after ischemic injury. Journal of Cerebral Blood Flow & Metabolism. 34 (3), 480-488 (2014).
- Cheng, X., et al. Dynamic Alterations of Brain Injury, Functional Recovery, and Metabolites Profile after Cerebral Ischemia/Reperfusion in Rats Contributes to Potential Biomarkers. Journal of Molecular Neuroscience. 70 (5), 667-676 (2020).
- Wang, X., et al. Dual role of intrauterine immune challenge on neonatal and adult brain vulnerability to hypoxia-ischemia. Journal of Neuropathology & Experimental Neurology. 66 (6), 552-561 (2007).
- Panahpour, H., Farhoudi, M., Omidi, Y., Mahmoudi, J. An In vivo Assessment of Blood-Brain Barrier Disruption in a Rat Model of Ischemic Stroke. Journal of Visualized Experiments. (133), e57156 (2018).
- Laake, J. H., Haug, F. M., Wieloch, T., Ottersen, O. P. A simple in vitro model of ischemia based on hippocampal slice cultures and propidium iodide fluorescence. Brain Research Protocols. 4 (2), 173-184 (1999).
- Cimarosti, H., Kantamneni, S., Henley, J. M. Ischaemia differentially regulates GABA(B) receptor subunits in organotypic hippocampal slice cultures. Neuropharmacology. 56 (8), 1088-1096 (2009).
- Cho, S., et al. Spatiotemporal evidence of apoptosis-mediated ischemic injury in organotypic hippocampal slice cultures. Neurochemistry International. 45 (1), 117-127 (2004).
- Dong, W. Q., Schurr, A., Reid, K. H., Shields, C. B., West, C. A. The Rat Hippocampal Slice Preparation as an Invitro Model of Ischemia. Stroke. 19 (4), 498-502 (1988).
- Tamura, R., et al. Neuroprotective effects of adenosine deaminase in the striatum. Journal of Cerebral Blood Flow and Metabolism. 36 (4), 709-720 (2016).
- Dennis, S. H., et al. Oxygen/Glucose Deprivation Induces a Reduction in Synaptic AMPA Receptors on Hippocampal CA3 Neurons Mediated by mGluR1 and Adenosine A(3) Receptors. Journal of Neuroscience. 31 (33), 11941-11952 (2011).
- Bar El, Y., Kanner, S., Barzilai, A., Hanein, Y. Activity changes in neuron-astrocyte networks in culture under the effect of norepinephrine. PLoS One. 13 (10), (2018).
- Kuszczyk, M. A., et al. Blocking the Interaction between Apolipoprotein E and A beta Reduces Intraneuronal Accumulation of A beta and Inhibits Synaptic Degeneration. American Journal of Pathology. 182 (5), 1750-1768 (2013).
- Skaper, S. D., Facci, L. Central Nervous System Neuron-Glia co-Culture Models and Application to Neuroprotective Agents. Methods in Molecular Biology. 1727, 63-80 (2018).
- Fang, A., et al. Effects of astrocyte on neuronal outgrowth in a layered 3D structure. BioMedical Engineering OnLine. 18 (1), 74 (2019).
- Fernando, G., Yamila, R., Cesar, G. J., Ramon, R. Neuroprotective Effects of neuroEPO Using an In vitro Model of Stroke. Behavioral Sciences. 8 (2), (2018).
- Zhao, L. R., Willing, A. Enhancing endogenous capacity to repair a stroke-damaged brain: An evolving field for stroke research. Progress in Neurobiology. 163, 5-26 (2018).
- Crandall, J. E., Jacobson, M., Kosik, K. S. Ontogenesis of microtubule-associated protein 2 (MAP2) in embryonic mouse cortex. Brain Research. 393 (1), 127-133 (1986).
- Chamak, B., Fellous, A., Glowinski, J., Prochiantz, A. MAP2 expression and neuritic outgrowth and branching are coregulated through region-specific neuro-astroglial interactions. Journal of Neuroscience. 7 (10), 3163-3170 (1987).
- Almeida, A., Medina, J. M. A rapid method for the isolation of metabolically active mitochondria from rat neurons and astrocytes in primary culture. Brain Research Protocols. 2 (3), 209-214 (1998).
- Bessa, A., et al. GPER: A new tool to protect dopaminergic neurons. Biochim Biophys Acta. 1852 (10), 2035-2041 (2015).
- De Simone, U., Caloni, F., Gribaldo, L., Coccini, T. Human Co-culture Model of Neurons and Astrocytes to Test Acute Cytotoxicity of Neurotoxic Compounds. International Journal of Toxicology. 36 (6), 463-477 (2017).
- Yang, L., Shah, K. K., Abbruscato, T. J. An in vitro model of ischemic stroke. Methods in Molecular Biology. 814, 451-466 (2012).
- Holloway, P. M., Gavins, F. N. Modeling Ischemic Stroke In Vitro: Status Quo and Future Perspectives. Stroke. 47 (2), 561-569 (2016).
- Rossi, D. J., Brady, J. D., Mohr, C. Astrocyte metabolism and signaling during brain ischemia. Nature Neuroscience. 10 (11), 1377-1386 (2007).
