Detection of Protein S-Acylation using Acyl-Resin Assisted Capture
Summary
Acyl-RAC (Acyl-Resin Assisted Capture) is a highly sensitive, reliable and easy to perform method to detect reversible lipid modification of cysteine residues (S-acylation) in a variety of biological samples.
Abstract
Protein S-acylation, also referred to as S-palmitoylation, is a reversible post-translational modification of cysteine residues with long-chain fatty acids via a labile thioester bond. S-acylation, which is emerging as a widespread regulatory mechanism, can modulate almost all aspects of the biological activity of proteins, from complex formation to protein trafficking and protein stability. The recent progress in understanding of the biological function of protein S-acylation was achieved largely due to the development of novel biochemical tools allowing robust and sensitive detection of protein S-acylation in a variety of biological samples. Here, we describe acyl resin-assisted capture (Acyl-RAC), a recently developed method based on selective capture of endogenously S-acylated proteins by thiol-reactive Sepharose beads. Compared to existing approaches, Acyl-RAC requires fewer steps and can yield more reliable results when coupled with mass spectrometry for identification of novel S-acylation targets. A major limitation in this technique is the lack of ability to discriminate between fatty acid species attached to cysteines via the same thioester bond.
Introduction
S-acylation is a reversible post-translational modification involving addition of a fatty acyl chain to an internal cysteine residue on a target protein via a labile thioester bond1. It was first reported as a modification of proteins with palmitate, a saturated 16-carbon fatty acid2, and therefore this modification is often referred to as S-palmitoylation. In addition to palmitate, proteins can be reversibly modified by a variety of longer and shorter saturated (myristate and stearate), monounsaturated (oleate) and polyunsaturated (arachidonate and eicosapentanoate) fatty acids3,4,5,6,7. In eukaryotic cells, S-acylation is catalyzed by a family of enzymes known as DHHC protein acyltransferases and the reverse reaction of cysteine deacylation is catalyzed by protein thioesterases, most of which still remain enigmatic8.
The lability of the thioester bond makes this lipid modification reversible, allowing it to dynamically regulate protein clustering, plasma membrane localization, intracellular trafficking, protein-protein interactions and protein stability9,10. Consequently, S-acylation has been linked to several disorders including Huntington’s disease, Alzheimer’s disease and several types of cancer (prostate, gastric, bladder, lung, colorectal), which necessitates development of reliable methods to detect this post-translational protein modification11.
Metabolic labeling with radioactive ([3H], [14C] or [125I]) palmitate was one of the first approaches developed to assay protein S-acylation12,13,14. However, radiolabeling-based methods present health concerns, are not very sensitive, time consuming, and only detect lipidation of highly abundant proteins15. A faster and nonradioactive alternative to radiolabeling is metabolic labeling with bioorthogonal fatty acid probes, which is used routinely to assay dynamics of protein S-acylation16. In this method, a fatty acid with a chemical reporter (alkyne or azide group) is incorporated into the S-acylated protein by a protein acyltransferase. Azide-alkyne Huisgen cycloaddition reaction (click chemistry) can then be used to attach a functionalized group, such as a fluorophore or biotin, to the integrated fatty acid allowing for detection of the S-acylated protein17,18,19.
Acyl-biotin exchange (ABE) is one of the extensively used biochemical methods for capture and identification of S-acylated proteins that bypasses some of the shortcomings of metabolic labeling such as unsuitability for tissue samples15. This method can be applied for analysis of S-acylation in a diverse range of biological samples, including tissues and frozen cell samples20,21. This method is based on selective cleavage of the thioester bond between the acyl group and the cysteine residue by neutral hydroxylamine. The liberated thiol groups are then captured with a thiol-reactive biotin derivative. The generated biotinylated proteins are then affinity-purified using streptavidin agarose and analyzed by immunoblotting.
An alternative approach termed acyl-resin assisted capture (Acyl-RAC) was later introduced to replace the biotinylation step with direct conjugation of free cysteines by a thiol-reactive resin22,23. This method has fewer steps compared to ABE and similarly can be used to detect protein S-acylation in a wide range of samples1.
Acyl-RAC consists of 4 main steps (Figure 1),
1. Blocking of free thiol groups;
2. Selective cleavage of the cysteine-acyl thioester bond with neutral hydroxylamine (HAM) to expose cysteine thiol groups;
3. Capturing of the lipidated cysteines with a thiol-reactive resin;
4. Selective enrichment of the S-acylated proteins after elution with reducing buffer.
The captured proteins can then be analyzed by immunoblotting or subjected to mass spectrometry (MS) based proteomics to assess the S-acylated proteome in a varied range of species and tissues22,24,25. Individual S-acylation sites can also be identified by trypsin digestion of the captured proteins and analysis of the resulting peptides by LC-MS/MS22. Here, we demonstrate how acyl-RAC can be used for simultaneous detection of S-acylation of multiple proteins in both a cell line and a tissue sample.
Protocol
Mice used in this protocol were euthanized according to NIH guidelines. The Animal Welfare Committee at University of Texas Health Science Center in Houston approved all animal work.
1. Preparation of cell lysates
- Prepare lysis buffer as described in Table 1. To 10 mL of PBS, add 0.1 g of n-dodecyl β-D-maltoside detergent (DDM) and rotate to dissolve. Add 100 µL of phosphatase inhibitor cocktail 2, ML211 (10 µM), PMSF (10 mM) and protease inhibitor cocktail (1x) and chill the buffer on ice before use.
- Transfer required media containing cells from the incubator into 15 mL or 50 mL conical tubes and spin cells at 350 x g for 5 min and aspirate to get rid of any cell debris.
NOTE: We used 1 x 107 cells for each acyl-RAC reaction to be performed. - Wash the pellet by resuspending it in 5 mL of PBS and spinning at 350 x g for 5 min.
NOTE: Perform step 1.3 quickly to avoid cell lysis due to extended incubation in PBS. - Add 600 µL of lysis buffer prepared in step 1.1 to the pellet and lyse it by shaking at 1500 rpm in a thermal shaker for 30 min at 4 °C.
- Clear lysates by centrifugation at 20,000 x g at 4 ˚C for 30 min to pellet detergent-insoluble materials. Collect cleared lysate in pre-cooled 1.5 mL microfuge tubes and keep them on ice.
- Perform a Bradford/BCA assay to estimate protein concentration. It is critical to ensure the same amount of protein across different samples prior to performing the experiment. We recommend using at least 500 µg of protein per reaction.
NOTE: In the experiments described, we used cultured (Jurkat) cells and primary splenocytes from mice tissue. The lysis method described above can be adopted to other cell types as well. The average protein concentration obtained for the abovementioned cell types is approximately 500 µg per 1 x 107 cells. Jurkat cells were maintained in RPMI-1640 medium modified to contain 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 4,500 mg/L glucose, and 1,500 mg/L sodium bicarbonate supplemented with 10% FBS at 37 °C with 5% CO2. We used 1 x 107 Jurkat cells per reaction. Primary splenocytes were isolated from mouse spleen tissue as described26. Briefly, spleen tissue was macerated on ice, followed by lysis of erythrocytes in hypotonic solution and separation from the lymphocytes by centrifugation. We used 1 x 107 primary cells per reaction.
2. Acyl-RAC: Blocking of free thiol groups
NOTE: All subsequent steps can be performed at room temperature (RT).
- Transfer lysate into a fresh 1.5 mL microfuge tube and perform chloroform-methanol (CM) precipitation as described below.
- Add methanol (MeOH) and chloroform (CHCl3) to the lysate at a final ratio of lysate: MeOH: CHCl3 of 2:2:1 and shake vigorously to create a homogeneous suspension.
- Spin at 10,000 x g for 5 min to form a pellet (“pancake”) at the interphase between aqueous and organic phases.
- Tilt the tube and aspirate as much solvent as possible using a needle or a gel loading tip.
- Air dry the protein pellet for a few minutes and gently wash it by adding 600 µL of MeOH and mixing gently to avoid breaking up the pellet
- Carefully remove the remaining MeOH and dry the protein pellet on a benchtop for approximately 5 min.
- (Optional) Perform an additional centrifugation step to spin down any broken pellet after the MeOH wash to avoid loss of sample.
NOTE: The experiment can be stopped after the CM precipitation step. Once the pellet is obtained, it may be stored in 500 µL of MeOH in -20 °C up to a week.- Dissolve the protein pellet in 200 µL of 2SHB buffer by vortexing at 42 °C/1500 rpm in a thermal shaker until the pellet is dissolved.
- (Optional) Incubate for an additional 5–10 min in a sonicating water bath to dissolve the pellet.
NOTE: The length of sonication varies depending on solubility of the material. However, prolonged sonication can cause protein degradation.
- Prepare 0.2% methyl methanethiosulfonate (MMTS) (v/v) in 2SHB by adding 2 µL of MMTS to 998 µL of 2SHB buffer.
NOTE: Use freshly prepared 0.2% MMTS for each experiment. - Add 200 µL of 0.2% MMTS in 2SHB to each tube to a final concentration of 0.1% MMTS. Incubate for 15 min at 42 ˚C with shaking at 1500 rpm in a thermal shaker.
3. Acyl-RAC: Hydroxylamine (HAM) cleavage and capture of S-acylated proteins
- Repeat 3-4x CM precipitations as described above to remove MMTS. Removal of MMTS can be estimated by the lack of distinct odor of MMTS. After each precipitation, dissolve the pellet in 100 µL of 2SHB buffer by vortexing at 42 ˚C/1500 rpm in a thermal shaker until the pellet dissolves, and then dilute with 300 µL of Buffer A.
- After final precipitation, dissolve samples in 200 µL of 2SHB buffer as described above and dilute with 240 µL of Buffer A.
- In case of comparing changes in S-acylation in response to several treatment conditions, measure protein concentration again and proceed with equal amount of protein for each condition.
- Retain 40 µL from each sample as an input control.
- Split samples into two equal parts of 200 µL and mark tubes as “+ HAM” and “- HAM”. Add 50 µL of freshly prepared neutral 2 M HAM (pH 7.0-7.5) to a final concentration of 400 mM to one of the tubes (+ HAM) and 50 µL of neutral 2 M NaCl to the second tube (- HAM), which will be used as a negative control. Proceed to addition of thiopropyl-Sepharose (TS) beads.
NOTE: The experiment can be stopped after any of the CM precipitation steps. Once the pellet is obtained, it may be stored in 500 µL of MeOH in -20 °C up to a week. Neutral pH of 2 M HAM ensures its selectivity for the acyl-cysteine thioester bond and should be carefully adjusted. Care should be taken when handling samples in 2SHB buffer to avoid loss of sample due to excessive foaming. All following steps are identical for – HAM and + HAM samples. - Add 30 µL of TS bead-slurry to each tube and rotate the tubes for 1-2 h at RT.
- Wash the TS beads 4x with 1% SDS in Buffer A to remove residual HAM.
- Gently spin down all bead samples using a microfuge for 1 min and carefully aspirate the supernatant.
- Resuspend the beads in 500 µL of 1% SDS in Buffer A.
- Repeat step 3.7.1–3.7.2 thrice.
NOTE: Activated thiopropyl-Sepharose (TS) is supplied freeze-dried in the presence of additives that must be washed away at neutral pH before coupling. Distilled water is recommended for swelling and washing. Weigh 0.1 g of beads and resuspend in 1 mL of distilled water and allow it to swell while rotating for 30 min-1 h at RT. Wash beads 1x with buffer A and prepare a slurry with buffer A, in a ratio of 50% settled medium to 50% buffer. Swollen TS may be stored at neutral pH in the presence of 20% ethanol at 4 °C up to a week. Do not use sodium azide as a bacteriostatic agent since azide ions react with the 2-pyridyl disulfide groups. For higher efficiency, prepare fresh beads. While handling the beads, the tip of a P200 pipette tip can be cut slightly so as to prevent any damage.
NOTE: The experiment can be stopped at any stage after CM precipitation. The pellet may be store in 500 µL of MeOH in -20 °C up to a week.
4. Elution and detection of S-acylated proteins
- After the last wash, gently spin down the beads as described above and aspirate as much supernatant as possible without disturbing the beads.
- Recover the proteins from beads with 4x SDS sample buffer with DTT.
- Add 50 µL of 4x SDS sample buffer to the beads and incubate at 80 ˚C, 1500 rpm for 15 min in a thermal shaker. Let the tubes cool.
- Centrifuge the beads at 5,000 x g for 3 min to completely pellet the beads and transfer the eluted proteins to a fresh 1.5 mL tube using a gel loading tip.
- Run SDS-PAGE and analyze S-acylation of the protein(s) of interest by western blotting.
Representative Results
Following the protocol described above, we first used acyl-RAC to simultaneously detect S-acylation of several proteins in Jurkat cells, an immortalized T cell line originally derived from the peripheral blood of a T cell leukemia patient27. Regulatory T cell proteins previously identified as S-acylated9,28,29 were chosen to demonstrate the utility of this method. As shown in Figure 2A, tyrosine kinase Lck can be detected in lysates treated with neutral 2 M hydroxylamine to cleave the thioester bond between cysteine residues and a fatty acid moiety (+ HAM lane). The sample treated with 2 M NaCl (- HAM lane) represents an important negative control, indicating the selectivity of the assay for detection of the thioester bonds. The input lane further confirms the specificity of the signal and can serve as a positive control if the protein of interest is not S-acylated. The membrane was then stripped and reprobed with antibodies against proteins Fyn and LAT to demonstrate that the acyl-RAC assay can be used to analyze S-acylation of multiple proteins at the same time (Figure 2A).
To demonstrate the utility of this method to detect protein S-acylation in tissue samples, we applied the acyl-RAC protocol to the mouse spleen. As shown in Figure 2B, S-acylation of Lck, Fyn and LAT can be readily detected in primary mouse splenocytes indicating that this modification is conserved between two species.
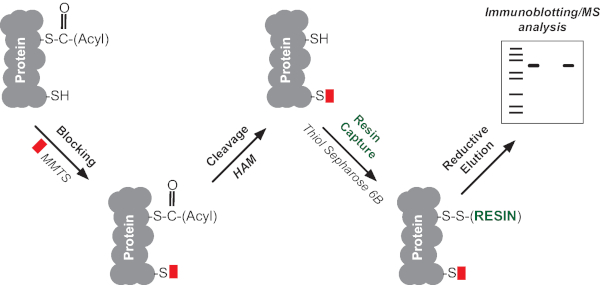
Figure 1. Schematic representation of acyl-RAC. The method consists of 4 main steps: (1) blocking free thiol groups with methyl methanethiosulfonate (MMTS), (2) selective cleavage of the cysteine-acyl thioester bond with neutral hydroxylamine (HAM) to expose cysteine thiol groups, (3) capture of proteins with newly exposed cysteine thiol by direct conjugation with a thiol-reactive resin and (4) recovery of captured proteins using a reducing elution buffer, followed by immunoblotting or MS analysis. Please click here to view a larger version of this figure.
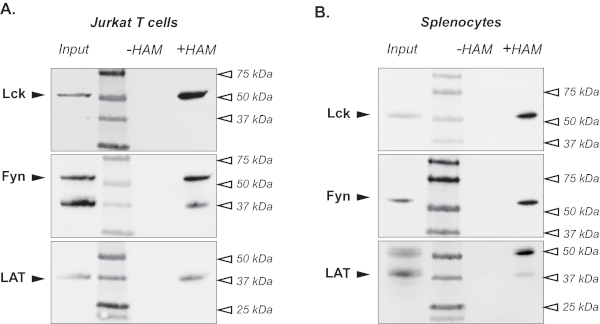
Figure 2. Detection of S-acylated proteins. An acyl-RAC assay was used to detect S-acylation of T cell signaling proteins in Jurkat T cells (A) and murine spleen tissue (B). Immunoblotting with Lck-, Fyn- and LAT-specific antibodies shows S-acylation of these proteins in the hydroxylamine treated sample (+ HAM lane). A sample treated with sodium chloride (- HAM lane) serves as negative control. Untreated lysates are shown in the input lane. Please click here to view a larger version of this figure.
| Buffer | Composition | Notes |
| Lysis Buffer (LB) | 1% DDM in DPBS; 10 µM ML211; Phosphatase Inhibitor Cocktail 2 (1:100); Protease Inhibitor Cocktail (1X), PMSF (10 mM) | Prepare fresh, chill at 4 ˚C before use. |
| SDS Buffer (2SHB) | 2% SDS; 5 mM EDTA; 100 mM HEPES; pH 7.4 | |
| Buffer A | 5 mM EDTA; 100 mM HEPES; pH 7.4 | |
| Hydroxylamine (HAM) | 2 M stock solution in distilled H20 | Prepare fresh. When first dissolved, HAM has a very low pH. Use 5 M NaOH to pH to 7-7.5. |
| 4X SDS sample buffer | 200 mM Tris-Cl (pH 6.8), 8% SDS (sodium dodecyl sulfate) 0.01% Bromophenol blue, 10% glycerol | Supplement with 5 mM DTT just before use. |
Table 1: Buffer composition of commonly used buffers required in the protocol. 2SHB buffer and Buffer A can be prepared in advance but their pH must be checked regularly. Lysis buffer and HAM must be prepared the day of experiment.
Discussion
Here, we successfully utilized the acyl-RAC assay to detect S-acylation of selected proteins in both cultured human cells and primary cells derived from mouse tissue. This method is simple, sensitive, and can be easily performed with minimal equipment requirements using standard biochemistry techniques. This method has been shown to successfully identify novel S-acylated proteins such as the β-subunit of the protein translocating system (Sec61b), ribosomal protein S11 (Rps11), and microsomal glutathione-S-transferase 3 (MGST3)22. The sensitivity and adaptability of acyl-RAC makes it suitable to study protein S-acylation in a variety of biological samples.
A significant fraction of S-acylated proteins can be associated with glycosphingolipid-enriched parts of the plasma membrane, also known as lipid rafts. Thus, some S-acylated proteins might not be fully extracted if mild detergents, such as Triton X-100, NP-40 or Brij58, are used for lysis30. Anionic detergents (such as SDS) or maltoside based non-ionic detergents (such as n-dodecyl β-D-maltoside (DDM)) can be used to ensure dissociation of lipid rafts and full recovery of the proteins of interest30,31,32. In this protocol, we used DDM, a mild, lipid-like non-ionic detergent that has been demonstrated to be effective in membrane protein extraction and maintaining their stable native state over prolonged periods33.
The unregulated thioesterase activity during lysis of the sample can potentially result in reduced protein yield after the TS pull-down step and thus affect the detection of S-acylated proteins. A thioesterase inhibitor, such as ML211, a dual inhibitor for Acyl-Protein Thioesterase 1 (APT1, LYPLA1) and Acyl-Protein Thioesterase 2 (APT2, LYPLA2) can be added to prevent indiscriminate deacylation of proteins in the lysate34,35.
An incomplete blockade of cysteine residues can cause binding of non-acylated proteins to TS, thus resulting in a high background evident by high signal in the –HAM samples. The background binding can be substantially reduced by an additional incubation with a thiol-crosslinking agent-2,2’-dithiodipyridine as described in a modified ABE protocol by Zhou et al.36.
In contrast to metabolic labeling, acyl-RAC is not limited to live cells and can be performed to detect protein S-acylation in freshly isolated or frozen tissue samples. Since the acyl-RAC protocol avoids an immunoprecipitation step, it can be used to simultaneously detect lipidation of multiple proteins of interest in the same assay, thus potentially reducing the amount of biological material required in a single experiment. However, metabolic labeling-based assays are more suitable for experiments aimed to specifically detect de novo S-acylation or measure fatty acid turnover rates on a protein of interest. Another important limitation of acyl-RAC is that this technique is unable to distinguish between different fatty acids that can be covalently bound to the cysteine residues via the same thioester bond3,4,5,6,7.
Both acyl-RAC and ABE assays are based on selective cleavage of the thioester bond between the cysteine residue and a fatty acid to reveal a free thiol group. Since acyl-RAC uses direct pull-down of S-acylated proteins by a thiol-reactive resin, this method requires fewer steps in comparison to ABE. Although similar, proteome-wide studies of S-acylation show some variation in the proteins detected using these two methods, most likely due to the technical differences. For example, in rat brain homogenate, ABE-based analysis identified 241 S-acylated proteins whereas acyl-RAC-based analysis identified 14425. 61 proteins in the rat brain proteome were commonly detected by both methods underscoring the need for use of multiple techniques to ascertain the S-acylation status of a protein.
Another potential drawback of acyl-RAC and ABE methods is the inability to reliably evaluate stoichiometry of S-acylation and determine the number of lipidated cysteines. A modification of these techniques, termed acyl-PEG exchange or PEG-shift assay, was developed to address these limitations37,38. In this method, pull-down step following hydroxylamine cleavage is substituted by incubation with a PEG-maleimide mass-tag. Resulting PEGylation of the exposed free cysteine residues can be observed as mass shift by SDS-PAGE.
In conclusion, acyl-RAC is a fast, sensitive and reliable method that can provide valuable insights into dynamics of protein S-acylation under both physiological and pathophysiological conditions in a great variety of biological samples. Considering limitations of the method discussed above, a combination of acyl-RAC with other techniques is recommended to fully characterize S-acylation of a protein of interest.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the National Institutes of Health grants 5R01GM115446 and 1R01GM130840.
Materials
| cOmplete Protease Inhibitor Cocktail tablets | Sigma | 11836170001 | |
| Eppendorf Centrifuge 5424 | Eppendorf | 22620444 | |
| Hydroxylamine (HAM) | Sigma | 159417 | |
| Methyl methanethiosulfonate (MMTS) | Sigma | 64306 | |
| Mini tube rotator | LabForce | ||
| ML211 | Cayman | 17630 | |
| Multi-Therm Cool-Heat-Shake | Benchmark Scientific | H5000-HC | |
| n-Dodecyl β-D-maltoside (DDM) | Sigma | D641 | |
| Phosphatase Inhibitor Cocktail 2 | Sigma | P5726 | |
| Thiopropyl-Sepharose 6B (TS) | Sigma | T8387 | |
| Ultrasonics Quantrex Sonicator | L & R |
References
- Bijlmakers, M. J. Protein acylation and localization in T cell signaling. Molecular Membrane Biology. 26 (1-2), 93-103 (2009).
- Magee, A. I., Courtneidge, S. A. Two classes of fatty acid acylated proteins exist in eukaryotic cells. EMBO Journal. 4 (5), 1137-1144 (1985).
- Fujimoto, T., et al. P-selectin is acylated with palmitic acid and stearic acid at cysteine 766 through a thioester linkage. Journal of Biological Chemistry. 268 (15), 11394-11400 (1993).
- DeMar, J. C., Anderson, R. E. Identification and quantitation of the fatty acids composing the CoA ester pool of bovine retina, heart, and liver. Journal of Biological Chemistry. 272 (50), 31362-31368 (1997).
- Montigny, C., et al. S -Palmitoylation and S -Oleoylation of Rabbit and Pig Sarcolipin. Journal of Biological Chemistry. 289 (49), 33850-33861 (2014).
- Muszbek, L., Laposata, M. Covalent modification of proteins by arachidonate and eicosapentaenoate in platelets. Journal of Biological Chemistry. 268 (24), 18243-18248 (1993).
- Hallak, H., et al. Covalent binding of arachidonate to G protein alpha subunits of human platelets. Journal of Biological Chemistry. 269 (7), 4713-4716 (1994).
- Tsutsumi, R., Fukata, Y. F. M. Discovery of protein-palmitoylating enzymes. Pflügers Archive: European Journal of Physiology. , 1206 (2008).
- Webb, Y., Hermida-Matsumoto, L., Resh, M. D. Inhibition of protein palmitoylation, raft localization, and T cell signaling by 2-bromopalmitate and polyunsaturated fatty acids. Journal of Biological Chemistry. 275 (1), 261-270 (2000).
- Paige, L. A., Nadler, M. J., Harrison, M. L., Cassady, J. M. Reversible palmitoylation of the protein-tyrosine kinase p56lck. Journal of Biological Chemistry. 268, 8669-8674 (1993).
- Blanc, M., et al. SwissPalm: Protein Palmitoylation database. F1000Research. 4, 1-23 (2015).
- O’Brien, P. J., Zatz, M. Acylation of bovine rhodopsin by [3H]palmitic acid. Journal of Biological Chemistry. 259 (8), 5054-5057 (1984).
- Drahansky, M., et al. We are IntechOpen , the world’s leading publisher of Open Access books Built by scientists. Intech. 13, (2016).
- Resh, M. D. Use of analogs and inhibitors to study the functional significance of protein palmitoylation. Methods. 40 (2), 191-197 (2006).
- Drisdel, R. C., Green, W. N. Labeling and quantifying sites of protein palmitoylation. Biotechniques. 36 (2), 276-285 (2004).
- Martin, B. R., Cravatt, B. F. Large-scale profiling of protein palmitoylation in mammalian cells. Nature Methods. 6, 135-138 (2009).
- Rami, N., Hannoush, N. A. -. R. Imaging the lipidome: omega-alkynyl fatty acids for detection and cellular visualization of lipid-modified proteins. ACS Chemical Biology. 4 (7), 581-587 (2009).
- Kostiuk, M. A., et al. Identification of palmitoylated mitochondrial proteins using a bio-orthogonal azido-palmitate analogue. FASEB Journal. 22 (3), 721-732 (2008).
- Charron, G., et al. Robust Fluorescent Detection of Protein Fatty-Acylation with Chemical Reporters. Journal of the American Chemical Society. 131 (13), 4967-4975 (2009).
- Roth, A. F., et al. Global analysis of protein palmitoylation in yeast. Cell. 125, 1003-1013 (2006).
- Kang, R., et al. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 456 (7224), 904-909 (2008).
- Forrester, M. T., et al. Site-specific analysis of protein S -acylation by resin-assisted capture. Journal of Lipid Research. 52 (2), 393-398 (2011).
- Guo, J., et al. Proteomic Profiling of Cysteine-Based Reversible Modifications. Nature Protocols. 9 (1), 64-75 (2014).
- Zaballa, M. E., van der Goot, F. G. The molecular era of protein S-acylation: spotlight on structure, mechanisms, and dynamics. Critical Reviews in Biochemistry and Molecular Biology. 53 (4), 420-451 (2018).
- Edmonds, M. J., Geary, B., Doherty, M. K., Morgan, A. Analysis of the brain palmitoyl-proteome using both acyl-biotin exchange and acyl-resin-assisted capture methods. Scientific Reports. 7 (1), 1-13 (2017).
- Lim, J. F., Berger, H., Su, I. H. Isolation and activation of murine lymphocytes. Journal of Visualized Experiments. (116), 1-8 (2016).
- Schneider, U., Schwenk, H., Bornkamm, G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. International Journal of Cancer. 19, 621-626 (1977).
- Bijlmakers, M. J. Protein acylation and localization in T cell signaling. Molecular Membrane Biology. 26 (1), 93-103 (2009).
- Hundt, M., et al. Palmitoylation-Dependent Plasma Membrane Transport but Lipid Raft-Independent Signaling by Linker for Activation of T Cells. Journal of Immunology. 183 (3), 1685-1694 (2009).
- Orwick-Rydmark, M., Arnold, T., Linke, D. The use of detergents to purify membrane proteins. Current Protocols in Protein Science. 84, 4810-4835 (2016).
- Brdicka, T., et al. Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. Journal of Experimental Medicine. 191 (9), 1591-1604 (2000).
- Akimzhanov, A. M., Boehning, D. Rapid and transient palmitoylation of the tyrosine kinase Lck mediates Fas signaling. Proceedings of the National Academy of Sciences of the United States of America. 112 (38), 11876-11880 (2015).
- Stetsenko, A., Guskov, A. An overview of the top ten detergents used for membrane protein crystallization. Crystals. 7 (7), (2017).
- Adibekian, A., et al. Optimization and characterization of a triazole urea dual inhibitor for lysophospholipase 1 (LYPLA1) and lysophospholipase 2 (LYPLA2). Probe Reports from the NIH Molecular Libraries Program. 1, 1-42 (2013).
- Dekker, F. J., et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nature Chemical Biology. 6, 449-456 (2010).
- Zhou, B., et al. Low-background acyl-biotinyl exchange largely eliminates the coisolation of non- s-acylated proteins and enables deep s-acylproteomic analysis. Analytical Chemistry. 91 (15), 9858-9866 (2019).
- Howie, J., et al. Substrate recognition by the cell surface palmitoyl transferase DHHC5. Proceedings of the National Academy of Sciences of the United States of America. 111 (49), 17534-17539 (2014).
- Percher, A., et al. Mass-tag labeling reveals site-specific and endogenous levels of protein S-fatty acylation. Proceedings of the National Academy of Sciences of the United States of America. 113 (16), 4302-4307 (2016).
