Successful purifications of motors and chassis structures were assayed by gel electrophoresis. SDS-PAGE analysis confirmed the successful extraction of dynein from yeast (Figure 2), as the final filtrate collected in step 2.3.7 showed a clear, sharp band at the position of ~350 kDa. As expected, this dynein band was absent from the flowthrough and wash that removed unwanted proteins, and the beads from which dynein was cleaved. The observation suggests that the IgG affinity purification and TEV protease cleavage were both highly efficient. Additionally, TEV protease was also present in the final filtrate and formed a clear band at ~50 kDa.
The successful MT affinity purification of dynein and kinesin proteins was also confirmed with SDS-PAGE analysis (Figure 3). While dynein showed up as a clear single band at ~350 kDa, kinesin showed up as slightly smearing multiple bands at ~120 kDa, possibly due to the presence of both phosphorylated and dephosphorylated forms of the protein14 and variable yields in oligo-labeling. A comparison between the dynein and kinesin bands before and after this MT affinity purification revealed a reduction in the motor concentration, as indicated by the decrease in the band intensity, which was likely due to the removal of non-functional motors. Despite the reduction, the concentrations of motors retained were sufficient for effective TIRF assays. In addition to the TEV protease, a noticeable amount of tubulin (~51 kDa) was present in the final supernatant, most likely due to the gradual decomposition of MTs during the experiment, or incomplete removal of the excess tubulin through centrifugation. However, the consistent motility of motor-chassis ensembles shown in TIRF assays suggests that tubulin and TEV protease did not interfere with motor functions (see Figure 6).
Folding of DNA origami structures was assayed by agarose gel electrophoresis. Figure 4 depicts the results of a gel analyzing the folding of a flexible segmented chassis with 2 motor binding sites. The shift in mobility between the pure unfolded scaffold strand (Lane 1) and the folding reaction (Lane 2) indicates origami folding. Additionally, the folding reaction in Lane 2 indicates the presence of some multimerization of chassis structures. Multimerization typically occurs, and requires subsequent purification of the well-folded monomeric structures. The unincorporated excess staples were also visible, displaying a high degree of mobility through the gel.
Folded origami reactions were purified to remove excess unincorporated staples and multimers of the chassis structure. Figure 5 shows the results of a glycerol gradient purification of a flexible chassis with 7 motor binding sites. The early fractions correspond to the low glycerol density at the top of the tube. They contain the excess staples. The late fractions correspond to the high glycerol densities at the bottom of the tube and contain multimers and aggregates of folded structures. In this gel, fraction 7 indicates a suitable fraction containing well-folded monomeric chassis. Note that the well-folded structure is isolated from both excess staples and multimers. While this gel is representative, and fraction 7 often contains useable chassis, each purification experiment yields slightly different results and fractions should always be assayed to determine which fraction is best for motility assays.
The motility of motor-chassis ensembles is easily detectable and measurable on the kymographs generated from TIRF movies. For instance, the kymographs (Figure 6) of flexible chassis conjugated to seven dynein proteins ("7D" ensembles) show highly processive runs at relatively consistent velocities, demonstrating that the ensembles were active and motile in the reconstituted system, and that MT affinity purification successfully removed most of the non-functional, immobile dyneins that could slow or stall the ensembles. The same TIRF experiment has been successfully performed on other chassis types with different compliance and motor numbers to reveal the effects of these factors on dynein teamwork8,9.
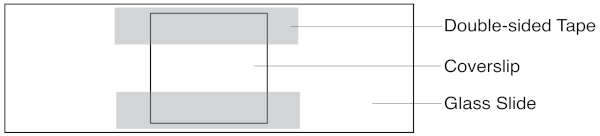
Figure 1: Schematic of the slide assay chamber. The coverslip sits atop two strips of double-sided tape. Solutions are pipetted in one side, and extracted with filter paper on the other. Please click here to view a larger version of this figure.
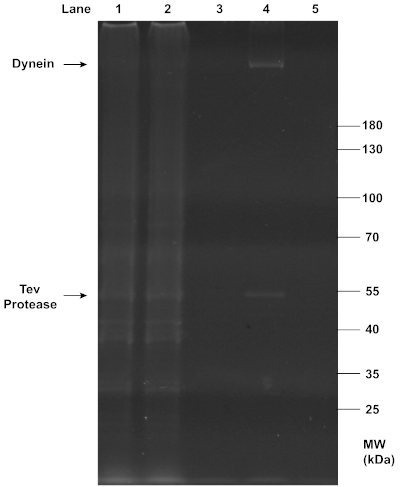
Figure 2: SDS-PAGE analysis of the purification of dynein from yeast. Yeast cells were lysed and centrifuged to collect the lysate (lane 1) containing total soluble proteins. The affinity between IgG on column beads and the ZZ tag on the recombinant dynein construct was exploited for this purification. The flow-through (lane 2) was collected from the mixture of the lysate and beads after it passed through a chromatography column. The dynein-bound beads were washed with buffers, and the first wash with the wash buffer (lane 3) was collected. Dynein was then conjugated to a DNA oligo. After TEV protease cleavage, centrifugation in a spin column separated the filtrate containing dynein (lane 4) and the residual beads (lane 5). All samples collected from the purification were denatured with 1x LDS Sample Buffer and 1x Reducing Agent, and loaded onto a 4-12% Bis-Tris gel. The gel was run in 1x MOPS buffer at 200 V for ~1 h, and stained with SYPRO Red Protein Gel Stain for imaging under UV light. Notably, the clear, sharp band at ~350 kDa in lane 4 indicates the presence of concentrated purified dynein in the final filtrate, while the band at ~50 kDa in the same lane indicates the co-presence of TEV protease. Please click here to view a larger version of this figure.
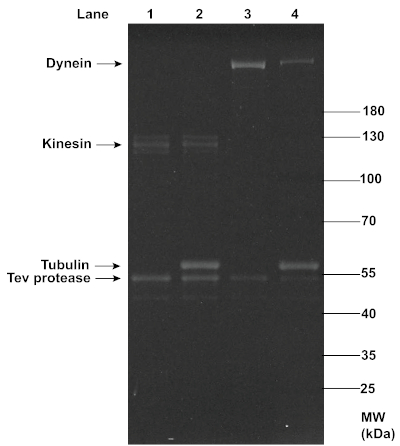
Figure 3: SDS-PAGE analysis of the MT affinity purification of functional dynein and kinesin proteins. Kinesin and dynein purified from yeast (lanes 1 and 3) were mixed with polymerized MTs and ATP, and ultracentrifugation was performed to separate the functional motors in the supernatants (lanes 2 and 4) from the non-functional motors that co-pellet with MTs. The motor samples before and after purification were denatured with 1x LDS Sample Buffer and 1x Reducing Agent, and loaded onto a 4-12% Bis-Tris gel. The gel was run in 1x MOPS buffer at 200 V for ~1 h, and stained with SYPRO Red Protein Gel Stain for imaging under UV light. The intensity of the motor bands (~120 kDa for kinesin, and ~350 kDa for dynein) appeared to decrease after the MT affinity purification, indicating a reduction in the motor concentrations. Noticeable concentrations of tubulin and TEV protease were present in the post-purification motor samples. Please click here to view a larger version of this figure.
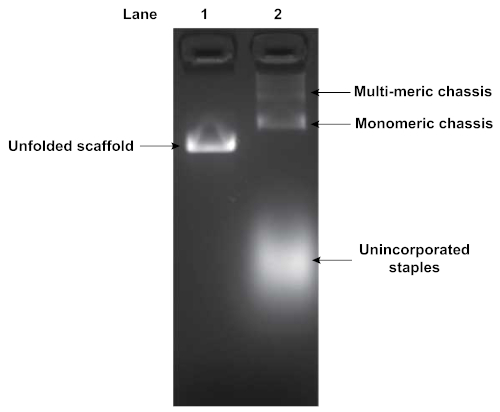
Figure 4: Agarose gel analysis of folded DNA origami structure. Folded DNA origami structures are assessed by gel electrophoresis. Lane 1 is the pure unfolded scaffold strand while lane 2 is the product of the folding reaction. Gel was run at 70 V in an ice water bath for 90 min in 0.5x TBE buffer supplemented with 11 mM MgCl2. A shift in mobility indicates origami folding. Unincorporated staples and chassis multimers were also visible in Lane 2. Please click here to view a larger version of this figure.
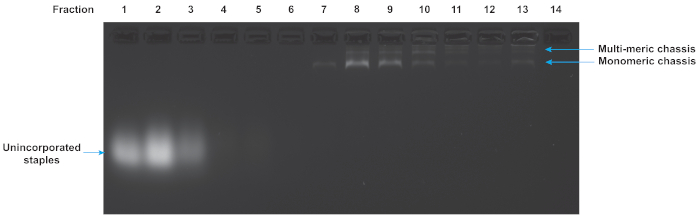
Figure 5: Agarose gel analysis of glycerol gradient purification of DNA origami chassis. The quality of the glycerol gradient purification can be determined by gel electrophoresis of the fractions from the centrifuge gradient. Low number fractions are from the top of the tube and correspond to low density of glycerol. High numbered fractions are from the bottom of the tube and correspond to high densities of glycerol. The excess staples, monomeric chassis, and multimeric chassis are all visible. Fraction 7 contains chassis suitable for TIRF microscopy as they are monomeric and excess staples are absent. Gel was run at 70 V in an ice water bath for 120 min in 0.5x TBE buffer supplemented with 11 mM MgCl2. Please click here to view a larger version of this figure.
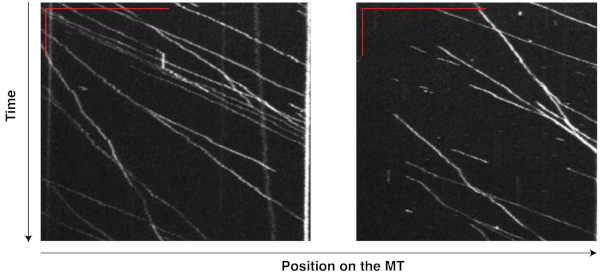
Figure 6: Kymographs showing the motility of dynein-chassis ensembles on single MTs. Dynein proteins extracted from yeast and purified with MTs were conjugated to flexible chassis structures. Each chassis had seven binding sites for dynein and formed a "7D" ensemble. The movement of these ensembles on MTs was recorded in a 10 min movie (200 ms exposure, 0.5 fps) during a TIRF assay. Kymographs from two MTs were generated from this movie in ImageJ by tracing along a single MT. The vertical and horizontal red bars in the top left corner of each image indicate 2 min and 20 µm, respectively. Each bright line records the movement of one ensemble, with the inverse of the slope indicating the velocity and horizontal displacement indicating the run length. With TIRF assays and kymography, the motility of motor-chassis ensembles becomes easily detectable and directly measurable. Please click here to view a larger version of this figure.
| Buffer Name | Composition | Step(s) Used | Comment |
| 5x Lysis Buffer | 150 mM HEPES (pH 7.4) | – | Filter sterilize the buffer. It can be stored at RT in a properly sealed container for a year. |
| 250 mM KAcetate | |||
| 10 mM MgAcetate | |||
| 5 mM EGTA (pH 7.5) | |||
| 50% glycerol | |||
| 4x Lysis Buffer With Supplements | 4x Lysis Buffer | 2.1-2.2 | Make this 4x buffer from the 5x lysis buffer above. Prepare a buffer without PMSF first, and add the compound (dissolved in pure ethanol) to a small aliquot of the buffer right before each step that requires it. |
| 4 mM DTT | |||
| 0.4 mM Mg-ATP | |||
| 2 mM PMSF | |||
| Wash Buffer | 1x Lysis Buffer With Supplements | 2.2 | To make the buffer, add KCl, Triton X-100, and ddH2O to the 4x lysis buffer with DTT and Mg-ATP, but do not add PMSF until right before use. |
| 250 mM KCl | |||
| 0.1% Triton X-100 | |||
| 5x TEV Buffer | 50 mM Tris-HCl (pH 8.0) | – | Filter sterilize the buffer. It can be stored at RT in a properly sealed container for a year. |
| 150 mM KCl | |||
| 10% Glycerol | |||
| 1x TEV Buffer | 1x TEV Buffer | 2.2-2.3 | To make the buffer, add DTT, Mg-ATP, Triton X-100, and ddH2O to the 5x stock TEV buffer, but do not add PMSF until right before use. |
| 1 mM DTT | |||
| 0.1 mM Mg-ATP | |||
| 0.1% Triton X-100 | |||
| 0.5 mM PMSF | |||
Table 1: Buffers for the IgG affinity purification of motor proteins from yeast cells (Protocol section 2).
| Buffer Name | Composition | Step(s) Used | Comment |
| 5x BRB80 | 400 mM PIPES | – | Filter sterilize the buffer. It can be stored at RT in a properly sealed container for a year. |
| 10 mM MgCl2 | |||
| 5 mM EGTA | |||
| Adjust pH to 6.8 with KOH | |||
| 1x BRB80 With Supplements | 1x BRB80 | 3.3 & 4.1-4.2 | Must be freshly made from the 5x BRB80 stock for every experiment. |
| 20 µM Taxol (dissolved in DMSO) | |||
| 1 mM DTT | |||
| 2x Polymerization Mix | 2x BRB80 (without supplements) | 3.3 | Flash freeze the mix in small aliquots and store at -80 oC. |
| 2 mM DTT | |||
| 2 mM Mg-GTP | |||
| 20% DMSO | |||
| Reconstitution Buffer | 1x BRB80 (without supplements) | 3.1 | Must be freshly made for every experiment. |
| 1 mM DTT | |||
| 1 mM Mg-GTP | |||
Table 2: Buffers for the polymerization of microtubules (Protocol section 3).
| Buffer Name | Composition | Step(s) Used | Comment |
| Taxol-Supplemented Lysis Buffer | 1x Lysis Buffer | 4.1 | Must be freshly made for every purification. |
| 20 µM Taxol (dissolved in DMSO) | |||
| 1 mM DTT | |||
| 5x ATP/Taxol Mix | 1x Lysis Buffer | 4.2 | Must be freshly made for every purification. |
| 25 mM Mg-ATP | |||
| 50 µM Taxol (dissolved in DMSO) | |||
| 5 mM DTT | |||
Table 3: Buffers for the microtubule affinity purification of functional motor proteins (Protocol section 4).
| Buffer Name | Composition | Step(s) Used | Comment |
| 20x Origami Folding Buffer | 100 mM Tris pH 8.0 | – | Can be stored at RT in a properly sealed container for up to a year. |
| 20 mM EDTA | |||
| 200 mM MgCl2 | |||
| 1x Origami Folding Buffer | 5 mM Tris pH 8.0 | 5.1-5.2 | Make the buffer fresh by diluting the 20x stock with ddH2O before every experiment. |
| 1 mM EDTA | |||
| 10 mM MgCl2 | |||
| 0.5x TBE | 45 mM TrispH 8.0 | 5.1-5.2 | Can be stored at RT in a properly sealed container for up to a year. |
| 45 mM Boric Acid | |||
| 1 mM EDTA | |||
Table 4: Buffers for the production of segmented DNA origami chassis (Protocol section 5).
| Buffer Name | Composition | Step(s) Used | Comment |
| DTT-Supplemented BRB80 | 1x BRB80 | 7.3 | Must be freshly made before every TIRF experiment. |
| 1 mM DTT | |||
| Taxol-Supplemented Lysis Buffer | 1x Lysis Buffer | 7.1 & 7.3 | Must be freshly made before every TIRF experiment. |
| 20 µM Taxol (dissolved in DMSO) | |||
| 1 mM DTT | |||
| Casein-Taxol-Supplemented Lysis Buffer | 1x Lysis Buffer | 7.1-7.3 | Must be freshly made before every TIRF experiment. |
| 20 µM Taxol (dissolved in DMSO) | |||
| 1 mM DTT | |||
| ~2.5 mg/ml Casein (dissolved in Tris-HCl at pH 8.0) | |||
| 1x Lysis Buffer | 30 mM HEPES (pH 7.4) | 7.2 | Make this buffer fresh by diluting the 5x stock (recipe detailed in Table 1) with ddH2O. |
| 50 mM KAcetate | |||
| 2 mM MgAcetate | |||
| 1 mM EGTA (pH 7.5) | |||
| 10% glycerol | |||
| 4x Energy Mix | 22.5 µL 1x Casein-Taxol-Supplemenfted Lysis Buffer | 7.3 | Must be freshly made before every TIRF experiment; volumes indicated are for a final volume of 25 µL. |
| 1 µL 0.1 M Mg-ATP | |||
| 1 µL 40% Glucose | |||
| 0.5 µL β-Mercaptoethanol | |||
| 4x Scavenger Mix | 24 µL 1x Casein-Taxol-Supplemented Lysis Buffer | 7.3 | Must be freshly made before every TIRF experiment; volumes indicated are for a final volume of 25 µL. |
| 1 µL 1x Oxygen Scavenger System | |||
Table 5: Buffers and mixes for the motor-ensemble motility TIRF assay (Protocol section 7). Details on the Oxygen Scavenger System used to make the Scavenger Mix can be found elsewhere17.
