Here we present a protocol that allows the analysis of protein:protein as well as protein:RNA complexes in phloem samples from Brassica napus. The workflow is illustrated in Figure 1. One major advantage of B. napus over other model organisms is the possibility of relatively easy phloem sample collection from small punctures in the inflorescence stem. This allows to obtain pure phloem samples in comparably large amounts. When sampling phloem, it is always advisable to check for contaminations, for example by RT-PCR8. A representative result obtained from a pure phloem sample is depicted in Figure 2. Non-contaminated phloem samples should show a visible band for thioredoxin h, whereas no signal for rubisco and the pollen coat protein should appear. The rubisco small subunit can serve as a control in samples from leaves, inflorescence stems or pollen. Thioredoxin h should be detectable in all samples, since its mRNA has been found in the phloem of different species, while the pollen coat protein transcript should only be visible in flower bud samples. Contaminations can occur when phloem sampling is performed at fully flowering plants (pollen coat protein) or when the first droplets from the phloem sampling punctures are not discarded properly (rubisco).
Prior to BN-PAGE it is mandatory to concentrate the phloem sap. Using 20 to 30 µg of protein per well, at least four major protein complex bands become visible after BN-PAGE of B. napus phloem sap, too low or too high concentrations do not allow a clear separation of the complexes (Figure 3). It is recommended to load several gel bands representing the same complex into one single pocket of the second-dimension gel to further concentrate the proteins from each single complex for downstream processing and subsequent mass spectrometric identification.
To identify the individual components of phloem macromolecular complexes, we combined the initial BN-PAGE with a second dimension Tris-Tricine-urea and a third dimension SDS-PAGE to achieve a separation of single proteins. Here, a separation with high resolution can be observed due to the different protein migration behaviors in urea and SDS-PAGE gels. Typically, proteins belonging to a single complex will be visible as a diagonal line across the gel. Proteins above this line have an increased hydrophobicity, whereas proteins below the line represent more hydrophilic ones28 (Figure 5, Figure 6).
For the characterization of RNP complexes, we used a part of a BN-PAGE gel to perform BN northern blotting. After blotting of a whole lane onto a nylon membrane and crosslinking by UV radiation, the RNA can be visualized using RNA-specific probes as illustrated in Figure 4. Here it is shown that a specific tRNA is only present in one specific complex. The protein components of this tRNA-binding complex can be further investigated using the 3D gel approach described above. Mass spectrometric analyses showed that this complex contains mainly tRNA-ligases what confirms the tRNA binding activity found by BN northern blotting.
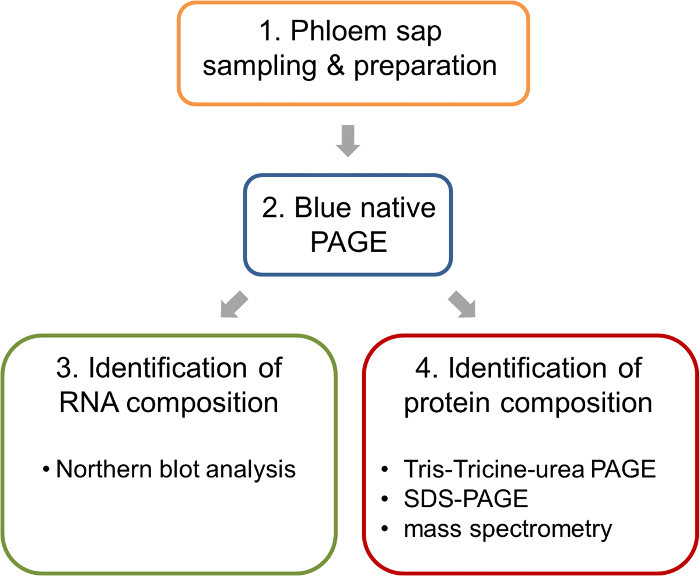
Figure 1: Work flow of the analysis of ribonucleoprotein complexes in the phloem sap of oilseed rape. After sampling and preparation of phloem samples, BN-PAGE is performed to separate native complexes. Such BN-PAGE gels allow the parallel analysis of nucleic acids by BN northern blotting and the identification of the protein components of the complexes by mass spectrometry. Please click here to view a larger version of this figure.
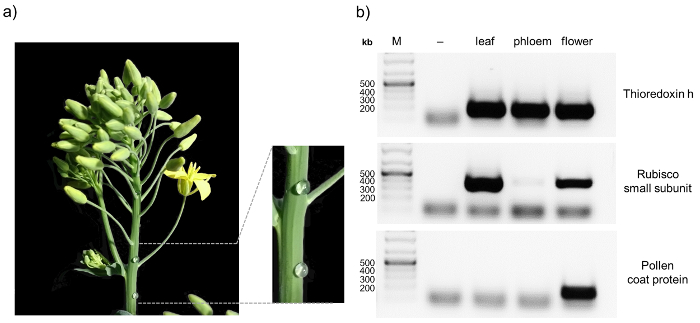
Figure 2: Sampling and purity control of phloem sap of B. napus. After puncturing the inflorescence stem of 8 – 10 week old oilseed rape plants with a sterile hypodermic needle, exudate is collected with a pipette and transferred to an ice-cold reaction tube (a) To confirm the purity of the phloem samples RT-PCR is performed, amplifying the transcripts of thioredoxin h, rubisco small subunit, and pollen coat protein in tissue-specific samples (b) RT-PCR without reverse transcriptase was performed as control (-). Please click here to view a larger version of this figure.
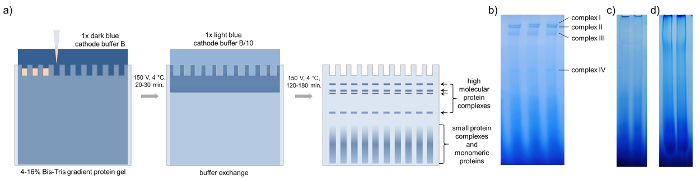
Figure 3: BN-PAGE. Schematic representation of blue native PAGE with concentrated phloem samples of B. napus. (a) After loading the gradient protein gel with 15-20 µL of concentrated phloem sap, BN-PAGE is performed with 1x dark blue cathode buffer B at 150 V for 20 – 30 min in a cold room. Then the buffer is exchanged against 1x light blue cathode buffer B/10 and the PAGE is finished at 150 V for 120 – 180 min until the dark blue running front runs out of the gel. The BN-PAGE gels show four distinct bands representing four different high-molecular-weight complexes. (b) Loading too little (c) or too much (d) phloem proteins reduces the visibility of individual complexes. Please click here to view a larger version of this figure.
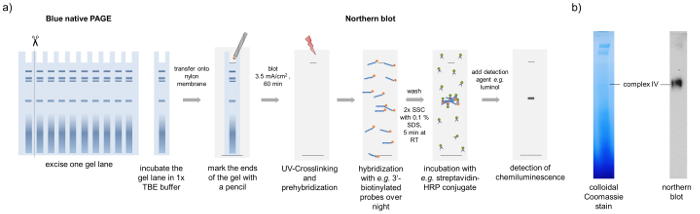
Figure 4: BN northern blotting. Schematic representation of the BN northern blotting method. (a) A lane of the BN-PAGE is transferred onto a nylon membrane by semi-dry blotting. After UV-crosslinking and pre-hybridization, the membrane is incubated with RNA specific probes (here a methionine tRNA-specific probe), which are biotinylated at the 3'-end in a hybridization oven over night. Free probes are removed by washing steps and the detection of the RNA is carried out by a streptavidin-HRP conjugate and luminol. Using this approach, we observed that complex IV from the BN-PAGE contains methionine tRNA. (b) The Coomassie stained gel and the tRNA blot resemble two different gel lanes run in parallel to avoid migration differences. Please click here to view a larger version of this figure.
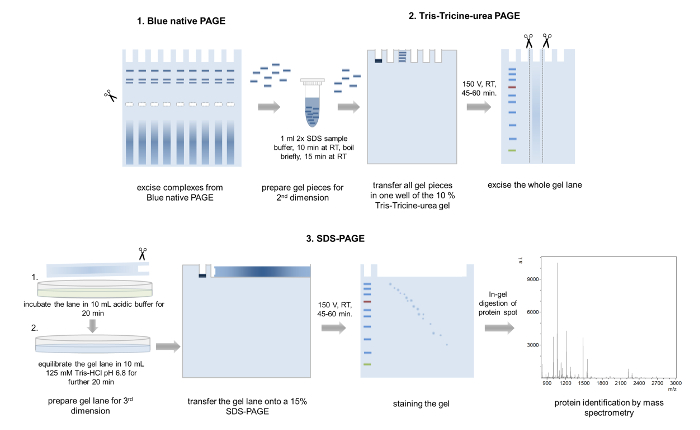
Figure 5: 3D-PAGE. Schematic representation of the 3D-PAGE approach. Several bands from the same complex are excised from a BN-PAGE gel, incubated in loading buffer and transferred into one well of the second dimension Tris-Tricine-urea-PAGE. After electrophoresis, whole gel lanes are cut out, washed in acidic acid buffer, equilibrated, and placed onto a 15% SDS-PAGE gel. After the third dimension PAGE, the separated proteins are Coomassie stained and analyzed by mass spectrometry. Please click here to view a larger version of this figure.
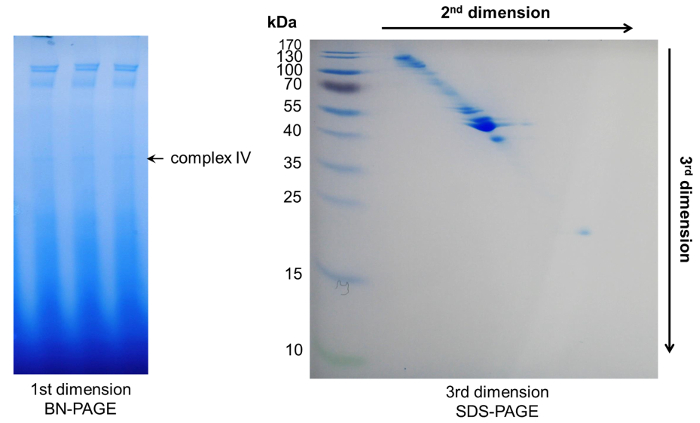
Figure 6: Representative results after 3D-PAGE. Within the first dimension BN-PAGE several complexes appeared. Their protein components could be further separated by two subsequent denaturing PAGEs, resulting in a diagonal spot pattern. Please click here to view a larger version of this figure.
| Probe | Sequence |
| Thioredoxin h | forward: CTCAAGGCAGCCAAAGAATC reverse: ATGGCCTGAACATCGAACTC |
| Rubisco small chain | forward: TTCACCGGCTTGAAGTCATC reverse: CCGGGTACTCCTTCTTGCAT |
| Pollen coat protein BRU77666 | forward: TCAGAACTGGAGCTTCAACG reverse: TTCCTTAATGGCCTCAGTGG |
Table 1: Primers used for phloem sample purity control. To confirm phloem sample purity primers specific for rubisco small chain, thioredoxin h, and pollen coat protein can be used for cDNA amplification.
| Buffers and Solutions | Content | Comment | |
| dark blue cathode buffer B | 15 mM Bis-Tris pH 7.0 50 mM Tricine 0.02% (w/v) Coomassie blue G250 |
recipe is adapted from Fiala et al. 17 pH should be adjusted to 7.0 Buffer can be made as a 10x stock |
|
| light blue cathode buffer B/10 | 15 mM Bis-Tris pH 7.0 50 mM Tricine 0.002% (w/v) Coomassie blue G250 |
Buffer can be prepared by diluting cathode buffer B with cathode buffer without Coomassie | |
| anode buffer | 50 mM Bis-Tris pH 7.0 |
Buffer can be prepared as a 10x stock solution | |
| 2x SDS sample buffer | 125 mM Tris-HCl pH 6.8 4% SDS 20% Glycerol 0.2 M DTT 0.02% (w/v) bromophenol blue |
||
| Transfer buffer 1x TBE buffer | 89 mM Tris pH 7.6 89 mM boric acid 2 mM EDTA |
TBE buffer can be made and stored as stocks of 5x or 10x. It is important to use RNase-free deionized water. | |
| 2x SSC buffer | 300 mM NaCl 30 mM Na3citrate pH 7.0 |
||
| 3x gel buffer | 3 M Tris 1 M HCl 0.3% SDS pH 8.45 |
recipe adapted from Schägger 19 | |
| Tris-Tricine gel solution A | For 10 mL: 30% acrylamide-bisacrylamide (29:1) 3.34 mL 6 M Urea 3x gel buffer 3.34 mL 70% glycerol 1.4 mL add ddH2O to 10 mL 10% APS 40 µL TEMED 4 µL |
Weight 6 M Urea and add 3x gel buffer, the acrylamide-bisacrylamide solution and 70% gycerol. Heat to 60 °C in a water bath to solubilize the urea. Add ddH2O to 10 mL, 10% APS and finally TEMED for polymerization. | |
| Tris-Tricine gel solution B | For 6 mL: 30% acrylamide-bisacrylamide (29:1) 0.8 mL 6 M Urea 3x gel buffer 1.5 mL add ddH2O to 6 mL 10 % APS 45 µL TEMED 4.5 µL |
Weight 6 M Urea and add 3x gel buffer and acrylamide-bisacrylamide solution. Heat to 60 °C in a water bath to solubilize the urea. Add ddH2O to 10 mL, 10% APS and finally TEMED for polymerization. | |
| 1x Tris-Tricine running buffer (anode) | 100 mM Tris 22.5 mM HCl pH 8.9 |
recipe adapted from Schägger 19 | |
| 1x Tris-Tricine running buffer (cathode) | 100 mM Tris 100 mM Tricine 1% SDS pH 8.25 |
recipe adapted from Schägger 19 | |
| Acidic buffer | 100 mM Tris 100 mM Acetic acid |
||
| 4% SDS stacking gel | For 2 mL: ddH2O 1.4 mL 30% acrylamide-bisacrylamide (37.5:1) 0.33 mL 1 M Tris pH 6.8 0.25 mL 10% SDS 20 µL 10% APS 20 µL TEMED 2 µL |
||
| 15% SDS separating gel | For 10 mL: ddH2O 2.3 mL 30% acrylamide-bisacrylamide (37.5:1) 5 mL 1 .5 M Tris pH 8.8 2.5 mL 10% SDS 100 µL 10% APS 100 µL TEMED 4 µL |
||
| 1x SDS running buffer | 25 mM Tris 192 mM glycine 0.1% SDS |
||
| Coomassie staining solution | 5% (w/v) aluminum sulfate-(14-18)-hydrate 10% (v/v) ethanol (96%) 2% (v/v) orthophosphoric acid (85%) 0.02% (w/v) Coomassie Brilliant Blue G-250 |
||
| ULTRAhyb Ultrasensitive Hybridization Buffer | Ambion, Life Technologies | ||
Table 2: Solutions and buffer recipes. List of all buffers and solutions necessary for 3D-PAGE analysis.
| Spot no. | MW obs. [kDa] | Identification | Organism | Accession no. | MW [kDa] | MASCOT score | |
| CIV_1 | 130 | Putative disease resistance protein At4g19050 | B. napus | CDX78917 | 131.1 | 110 | |
| CIV_2 | 130 | Putative disease resistance protein At4g19050 | B. napus | CDX78917 | 131.1 | 89 | |
| CIV_3 | 100 | Heat shock 70 kDa protein 14-like | B. napus | XP_013748865 | 89.9 | 113 | |
| CIV_4 | 90 | Eukaryotic elongation factor 2 Cell division control protein 48 homolog A |
B. napus B. napus |
CDX90241 CDY41316.1 |
89.5 89.5 |
111 197 |
|
| CIV_5 | 85 | Heat shock protein 90-2-like | B. rapa | XP_009132342 | 79.8 | 172 | |
| CIV_6 | 80 | Threonine–tRNA ligase Glycine–tRNA ligase |
B. oleracea B. napus |
XP_013599115 CDY43195 |
81.5 80.8 |
110 88 |
|
| CIV_7 | 70 | Heat shock protein 70 kDa Lysine–tRNA ligase-like |
B. oleracea B. napus |
XP_013685267 XP_013747530 |
67.8 69.9 |
230 150 |
|
| CIV_8 | 65 | Myrosinase Aspartate–tRNA ligase 2 Asparagine–tRNA ligase 1 |
B. napus B. napus B. napus |
ABQ42337 XP_013670803 XP_013729126 |
60.6 61.6 63.5 |
183 141 153 |
|
| CIV_9 | 60 | Myrosinase | B. napus | ABQ42337 | 60.6 | 164 | |
| CIV_10 | 55 | Adenosylhomocysteinase 2-like | B. rapa | XP_009135865 | 53.1 | 139 | |
| CIV_11 | 55 | Elongation factor 1-alpha 1-like | B. oleracea | XP_013586115 | 51.9 | 161 | |
| CIV_12 | 50 | Cystine lyase CORI3-like | B. napus | XP_013653143 | 48.1 | 237 | |
| CIV_13 | 44 | Cystine lyase CORI3-like | B. rapa | XP_009108611 | 48.3 | 213 | |
| CIV_14 | 38 | Fructose-bisphosphate aldolase | B. rapa | XP_009115993 | 38.4 | 226 | |
| CIV_15 | 45 | Cystine lyase CORI3-like | B. rapa | XP_009108611 | 48.3 | 219 | |
| CIV_16 | 45 | Cystine lyase CORI3 | B. rapa | CDY69765 | 46.9 | 209 | |
| CIV_17 | 38 | Fructose-bisphosphate aldolase | B. rapa | XP_009115993 | 38.4 | 124 | |
| CIV_18 | 18 | Peptidyl-prolyl cis-trans isomerase CYP18-3-like | B. napus | XP_009101932 | 18.3 | 128 | |
| CIV_19 | 45 | Cystine lyase CORI3-like | B. rapa | XP_009108611 | 48.3 | 177 | |
Table 3: Identified protein components from complex IV. Several tRNA ligases and further proteins have been found within the tRNA binding complex using MALDI-TOF mass spectrometric analysis after 3D-PAGE.
