All animal procedures were approved by the University of Cambridge Animal Welfare and Ethical Review Body and conformed to the Animals (Scientific Procedures) Act 1986 Amendment Regulations (SI 2012/3039).
1. Preparation in Advance
- Place an aliquot of basement membrane matrix (BMM) (~200 µL) on ice to thaw.
NOTE: The BMM solidifies at room temperature (RT). - Pre-warm 50 mL sterile high-glucose Dulbecco's Modified Eagle Medium (DMEM) (with no additions) and ~40 mL sterile culture medium (high-glucose DMEM supplemented with 10% Fetal Bovine Serum (FBS), 100 U/mL penicillin and 0.1 mg/mL streptomycin (P/S), and 2 mM L-glutamine) in 50 mL centrifuge tubes in a water bath at 37 oC.
- Pre-chill calcium and magnesium-containing Phosphate-Buffered Saline (PBS).
- Weigh out 15 mg collagenase (XI crude), add to a 50 mL centrifuge tube and keep it on ice.
- CAUTION: Collagenase is harmful. It causes skin irritation (H315) and serious eye irritation (H319). It may cause allergy or asthma symptoms or breathing difficulties if inhaled (H334). It may cause respiratory irritation (H335). Use appropriate personal protective equipment, avoid dust formation and avoid breathing dust. Ensure adequate ventilation.
2. Tissue Collection
- Add ~20 mL L-15 medium to a 50 mL centrifuge tube and place it on ice.
- Euthanize a mouse by cervical dislocation, or other approved schedule 1 method.
NOTE: Intestinal tissue is typically obtained from mice on a C57BL6 background. However, other genetic backgrounds have also been used e.g. 129/SvEv15. Imaging experiments require the use of fluorescent reporter mice e.g. GLU-Venus1. Tissue is obtained from adult mice (2-6 months) of both sexes. The mice are housed in individually-ventilated cages with ad libitum access to water and regular chow. However, experiments to test the effect of a high-fat diet, for example, on enteroendocrine cell function16 can be performed if supported by a project license. - Dissect and gently remove mouse intestine (from pylorus to start of rectum) using forceps and dissection scissors. Store it in L-15 medium on ice until ready to use.
3. Tissue Preparation
- Place intestinal tissue in a 10 cm petri dish containing enough PBS to cover the tissue. Take 10 cm of desired tissue (e.g. upper small intestine, top 10 cm, distal to pylorus).
- Flush out intestinal contents using a plastic Pasteur pipette and chilled PBS.
- Using forceps, delicately grip one end of the intestinal segment and place the tip of a Pasteur pipette into it. Flush out contents with chilled PBS. Repeat from both ends until the majority of the contents has been flushed out. Transfer to a clean petri dish containing fresh chilled PBS.
- Using forceps, remove the adipose tissue and mesentery, taking care to not pull off the muscle layer at the same time.
- Peel the muscle layer off "like a sock" under a dissecting microscope using two sets of fine forceps.
NOTE: This step is not essential but is highly recommended. There are alternative ways of removing the muscle layer to the one described here. For example, the intestine can be cut open longitudinally first, prior to the removal of the muscle layer.- Find a starting point at the more proximal end of the tissue where there is a visible flap of muscle. Gently pull away a small amount of muscle layer all the way around the intestine.
NOTE: To avoid tearing of the muscle layer or the intestinal epithelium, reduce the tension force by clamping a larger surface area rather than using the tips of the fine forceps. - Clamp the intestine and as much of the muscle flap as possible, gently pull apart and start peeling off the muscle layer from around the intestine. To avoid tearing both the muscle layer and epithelium, keep readjusting the position of the forceps to keep them close together. In this way, remove the muscle layer from the entire length of the intestinal segment and discard.
- Find a starting point at the more proximal end of the tissue where there is a visible flap of muscle. Gently pull away a small amount of muscle layer all the way around the intestine.
- Cut the intestine open longitudinally and wash by swirling in a clean petri dish with fresh chilled PBS. Repeat if necessary to remove any remaining chyme or mucus.
- Mince tissue with a surgical scalpel blade to achieve squares of ~1-2 mm2 and add these to ~20 mL chilled PBS in a 50-mL centrifuge tube using a Pasteur pipette. To avoid tissue pieces sticking to the pipette, cut off the tip and wet the pipette by triturating with PBS.
- Gently shake the tube to further wash the tissue pieces. Allow the tissue to settle and pour or pipette off the majority of the PBS and repeat with fresh PBS until the PBS looks clear.
4. Preparation of BBM-coated Plate/Dishes and Digestion Medium
NOTE: The following steps should be performed in a tissue culture hood (with incubation steps in 37 °C water bath).
- Prepare a 2% BMM solution in chilled DMEM (with no additions). While working, keep the solution on ice. For a 2% solution, add 140 µL thawed BMM to 7 mL DMEM (enough to prepare a single 24-well plate).
- Add 250 µL 2% BMM solution per well (24-well plate) or per glass bottom imaging dish.
- Incubate coated plates/dishes for at least 30 min in an incubator at 37 °C to allow for adequate polymerization of the BMM.
- Add the 50 mL pre-warmed DMEM (with no additions) to the 15 mg collagenase (from Steps 1.2 and 1.4) to form a 0.3 mg/mL solution and invert to dissolve.
- Once completely dissolved, using a 20 mL syringe, filter the collagenase solution through a 0.2 µm sterile filter into a new sterile 50 mL centrifuge tube. Label this as the digestion medium.
5. Tissue Digestion
- Remove the tissue pieces from the PBS using a 10 mL serological pipette and add to a sterile 50-mL centrifuge tube containing chilled sterile DMEM (with no additions), swirl and then remove DMEM.
NOTE: To avoid tissue sticking to the serological pipette, wet it by trituration with DMEM prior to contact with the tissue. - 'Digest' 1 and 2: Removal of cell debris and single cells
- Add 7 mL digestion medium to the tissue pieces and give the tube a swirl.
- Incubate for 5 min in a water bath at 37 °C.
- Gently shake (not swirl) the tube for ~3 s.
- Allow the tissue to settle and discard the digestion medium, reserving a small volume to look at under the microscope.
NOTE: Especially when starting out, it is highly recommended to inspect a small volume of each 'digest' (~30 µL) under the microscope to gauge the progress of the digestion process. If many crypts are observed at this stage, the shaking may be too vigorous. For representative images of what the different 'digest' stages typically look like, refer to Figure 1. - Repeat Steps 5.2.-5.2.4 (with fresh digestion medium).
- 'Digests' 3 to 5: Collection of crypt fragments
- Add 7 mL digestion medium to the tissue.
- Incubate for 10 min at 37 °C.
- During the incubation, shake every 5 min for 10-12 s.
NOTE: The shaking is more vigorous than the shaking for 'digests' 1 and 2.
- During the incubation, shake every 5 min for 10-12 s.
- Allow undigested tissue to settle and collect the digestion medium in a 15 mL centrifuge tube. If tissue is accidentally collected along with the medium, allow it to settle, remove it using a 10 mL serological pipette and transfer it back to the 50-mL centrifuge tube containing the undigested tissue.
- Centrifuge collected medium/supernatant at RT for 3 min at 100 x g.
- Discard the supernatant and re-suspend the cell pellet in 5 mL pre-warmed culture medium by gentle trituration and set aside.
- Inspect a small volume of the cell suspension under the microscope (see Note from Step 5.2.4).
NOTE: Ideally, crypt fragments start to appear in 'digest' 3 along with cell debris and single cells. 'Digests' 4 and 5 contain a significantly greater number of crypt fragments with reduced cell debris (Figure 1). Adjust shaking intensity as necessary i.e. if the tissue is not digesting and crypt fragments are not appearing, shake more vigorously. - Repeat steps 5.3.1-5.3.6 until 5 'digests' have been completed or until the majority of the tissue has been digested (a 6th digest may be required).
- Once all supernatants from 'digests' 3-5 (or 3-6) have been collected, centrifuge the digest supernatants at RT for 3 min at 100 x g.
- For secretion experiments, combine the supernatants from 'digests' 3-5 (or 3-6) prior to centrifugation.
NOTE: Less tissue is needed for imaging experiments, therefore choose the digest supernatant that is the cleanest (absence of cell debris and single cells) with the greater number of crypt fragments. This is typically 'digest' 4 or 5. - Discard the supernatant and gently re-suspend the pellet by triturating until no clumps are visible. Re-suspend the pellet in pre-warmed culture medium supplemented with 10 µM Y-27632 dihydrochloride (to prevent anoikis17). Use 5 mL for secretion experiments and 2 mL culture medium for imaging experiments.
- Filter cell suspension through a 100-µm filter (to remove any undigested tissue). Run a further 2 mL of pre-warmed culture medium through the filter to wash (total volume: 7 mL and 4 mL for secretion and imaging, respectively).
NOTE: Filtering is not essential, however, larger tissue fragments tend not to adhere to the plate/dishes.
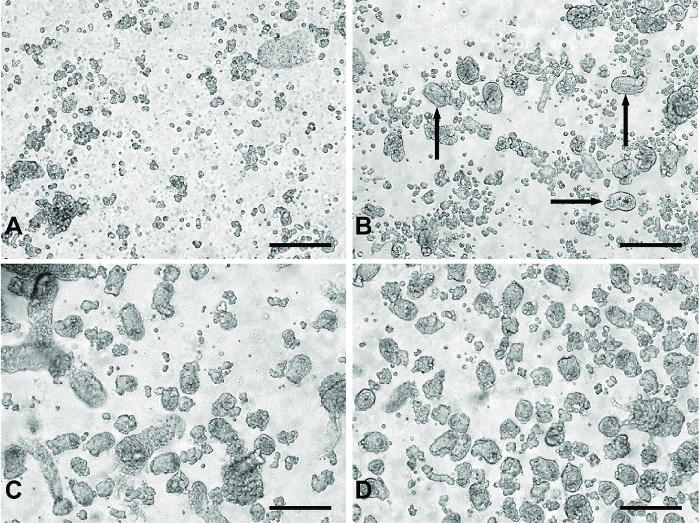
Figure 1: Representative images of 'digests' from primary small intestinal culture method. (A) Typical material from 'digests' 1 and 2 containing primarily single cells and cell debris. (B) An example of products from 'digest' 3. The black arrows indicate crypt fragments appearing in the digest material. (C) Typical of 'digest' 4 or 5. Panel (D) represents pooled digest material from 'digests' 3-5 passed through a 100 µm filter to remove the larger unwanted tissue fragments seen in (C). All images were taken using a digital inverted microscope with a 20X objective. Scale bars = 100 µm. Please click here to view a larger version of this figure.
6. Plating Intestinal Cultures (Enriched for Crypt Cells)
- Remove excess BMM solution from plate or dishes, and add 250 µL pre-warmed culture medium per secretion well.
NOTE: Imaging dishes are left without medium so proceed to the next step swiftly to prevent the dish from drying out. - Plate 250 µL cell suspension per well (24-well plate) or per glass bottom dish.
- Plate drop-wise in a slow 'zig-zag' motion across the well. Allow crypt fragments to settle for ~5 min before moving the plate to the incubator.
NOTE: This should encourage an even distribution of crypts/cells within the well.
- Plate drop-wise in a slow 'zig-zag' motion across the well. Allow crypt fragments to settle for ~5 min before moving the plate to the incubator.
- Incubate plate or dishes overnight at 37 °C and 5% CO2.
NOTE: A 'patchy' monolayer should have formed. For a representative image of cultures following three washes, see Figure 2A. - 'Flood' imaging dishes with 2 mL pre-warmed culture medium per dish.
NOTE: These dishes will be suitable for imaging for up to ~72 h following plating. Colonic cultures typically last longer, up to 7 days. Cells which have not adhered can be removed by a wash step. Primary cultures are now ready to be used for experiments.
The use of serial digestion steps allows the collection of a relatively clean preparation of crypt fragments to be plated for experiments. The first two digests remove primarily single cells and cell debris from the digesting material (Figure 1A). During the third digest, crypt fragments appear in the digested material (Figure 1B). Digests 4 and 5 yield a greater number of crypt fragments with fewer single cells (Figure 1C). Filtering the pooled material from digests 3-5 removes larger pieces of undigested tissue, which may be detrimental to the culture, producing a clean crypt fragment preparation (Figure 1D).
Following 18-24 h of culture, monolayers of primary small intestinal cells are observed (Figure 2A). Using cultures generated from transgenic mice specifically expressing the calcium fluorescent sensor, GCaMP3, under the control of the proglucagon promoter11, L cells are readily identifiable and interspersed within the culture (Figure 2B). In primary small intestinal cultures, compounds targeting the Gq-Ca2+i and cAMPi-dependent pathways stimulated the secretion of GLP-1 (for secretion experiment methodology see Reimann et al.1). Bombesin (BBS, 100 nM) and co-application of forskolin and 3-isobutyl-1-methylxanthine (IBMX) (F/I, 10 µM each) triggered a 2- and 11-fold, stimulation of GLP-1 release relative to basal, respectively (Figure 2C). Specific L cell expression of GCaMP3 allows the identification of and real-time monitoring of calcium mobilization in individual L cells. Both 100 nM bombesin (BBS) and 30 mM potassium chloride (KCl) stimulated transient increases in intracellular calcium indicative of the known Gq– and electrogenic-coupled pathways in primary L cells (Figure 2D).
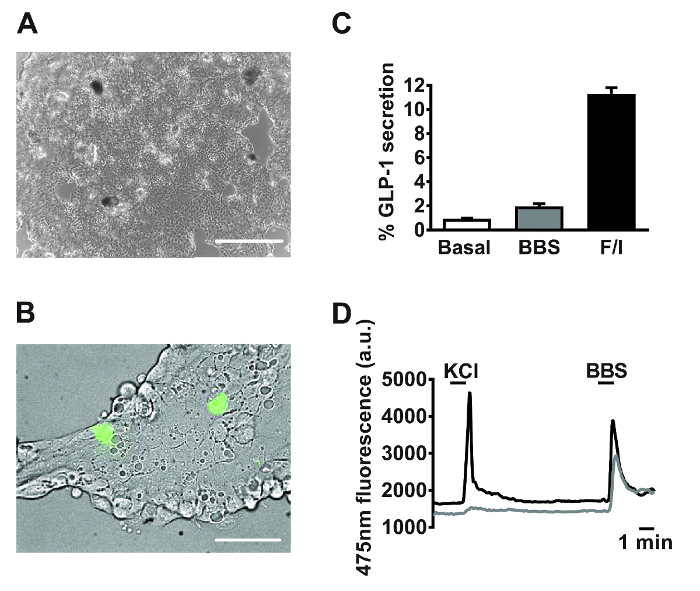
Figure 2: Representative data derived from primary small intestinal cultures. Example images of mixed primary small intestinal cultures used for (A) secretion and (B) imaging experiments post 24 h plating. (A) Image taken from a 24-well plate, using a digital inverted microscope with a 4X objective. Scale bar = 500 µm. (B) Using GLU-Cre x ROSA26 GCamP3 mice, L cells in the field of view (green cells) were identified by the fluorescence of GCaMP3. Scale bar represents 50 µm. (C) GLP-1 secretion was measured in response to bombesin (BBS, 100 nM) and forskolin/3-isobutyl-1-methylxanthine (IBMX) (F/I, 10 µM each). Percentage GLP-1 secretion was calculated by measuring GLP-1 levels in the supernatants and cell lysates. Data represent the means ± SEM of n = 3 for each condition. (D) GCaMP3 fluorescence (reflecting cytosolic calcium) of the two cells outlined in (B) monitored in real time in response to potassium chloride (KCl, 30 mM) and BBS (100 nM). Single cell imaging was performed using an inverted fluorescence microscope with a 40X oil-immersion objective. GCaMP3 was excited at 475/10 nm, using a 75 W xenon arc lamp and a monochromator controlled by fluorescence imaging software. Emission was recorded with a high-resolution digital charge-coupled device (CCD) camera using a dichroic mirror and a 510-560 nm bandwidth filter. Please click here to view a larger version of this figure.
