1H and 13C NMR spectra were collected for commercially available fish oil supplements using two NMR instruments; an 850 MHz and a 500 MHz spectrometer. These spectra can be used for the quantitative determination of components of fish oil, such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), as well other compounds such as n-1 acyl chains and nutritionally important index such as the n-6/n-3 ratio. The quantification can be performed even without the use of an internal standard, however, the quantitative results must be expressed as relative molar percentages. When the data need to be expressed in absolute values (mg/g), an internal standard is required. The results obtained by NMR are highly reproducible with relative standard deviations (RSD) ranging from 0.3% to 2% for 13C NMR analysis and from 0.5% to 2.5% for 1H NMR analysis, depending on the lipid. The slightly higher RSD for 1H NMR is often observed because proton spectra tend to be overcrowded, which affects the accuracy of the analysis, especially for resonances that have a lower signal to noise ratio (S/N). A very good agreement was found between the 850 MHz and the 500 MHz instrument with RSDs ranging from 1% to 4%. Relatively high RSDs (up to 8%) were observed when comparing results obtained by 1H and 13C, especially for compounds that appear in lower concentrations such as n-1 acyl chains. NMR spectroscopy has been previously validated as a tool for lipid analysis, including the determination of some fish oil components. Results showed that it is in good agreement with traditional methods, such as GC17,18.
1H NMR Analysis
Figure 1 compares the 1H NMR spectra acquired on (A) an 850 MHz and (B) a 500 MHz instrument. The 850 MHz spectrum is characterized by higher resolution, however the major components of fish oil including DHA, EPA, and n-6/n-3 ratio can also be determined from the 500 MHz spectrum. The 1H-NMR signals of fish oil fatty acids that can be used for quantitation purposes are shown in Table 1, whereas the complete NMR assignment of the 1H NMR spectrum of fish oil can be found elsewhere19.
1H NMR gave reliable data for the quantification of the total amount of n-3, n-6, DHA, trans fatty acids, n-1 acyl chains, and saturated fatty acids (SFA). For the 1H NMR analysis, the use of appropriate relationships is required because most of the signals belong to groups of protons that are common to different fatty acids and lipids. For that reason, in most cases the concentration of fatty acids in fish oil can be determined only by combination of various 1H NMR signals, incorporated in the appropriate relationships. In addition, these equations contain arithmetic coefficients that normalize the different number of protons associated with each group. When an internal standard is used the following equation should be considered: C = I/IIS × NIS/N × A × MW/m (1), where C is the concentration of the analyte in mg/g of fish oil, I is the integral of a resonance that is uniquely attributed to the lipid of interest, IIS is the area of a proton signal that belongs uniquely to the internal standard, N is the number of protons of the functional group that is analyzed, NIS is the number of protons of the internal standard that are used for the analysis, A is the millimoles of internal standard, MW is the molecular weight of the fatty acid (expressed in methyl esters), and m is the amount of fish oil expressed in g.
Example 1, DHA: The proportion of DHA is determined by the equation CDHA = ¾ IDHA/S, where IDHA is the integral of the signal at δ 2.39 which belongs to the Hα and Hβ protons of DHA, and S is the sum of integrals of the methyl protons of SFA, n-6, n-9, n-3, trans fatty acids plus the integrals of the peaks of n-1 acyl chains at δ 4.98, δ 5.05 and δ 5.81. The integral IDHA is normalized by multiplying by 3/4 because it corresponds to four protons, whereas the integral S corresponds to three protons. 1H NMR is not capable to giving information about positional distribution of fatty acids on the glycerol backbone and thus can only be used for the quantification of the total amount of fatty acids. The 1H NMR analysis of an encapsulated fish oil supplement showed that it consists of 10.5% of DHA. The concentration of DHA in the same sample using BHT was found to be 105.23 mg/g. These values are very close to the values obtained with 13C NMR (see example 2 for 13C analysis).
Example 2, n-1 acyl chains: The concentration of n-1 acyl chains is given by the relationship Cn-1 = 3In-1/S, where In-1 is the integral of the signal at δ 5.818. This signal corresponds to one proton and thus needs to be normalized by multiplying by three. When using BHT, n-1 acyl chains are determined by the equation Cn-1 = 2In-1/IBHT. The results cannot be expressed in mg/g because the MW of n-1 acyl chains is unknown.
Example 3, n-6/n-3 ratio: This important index can be calculated from the ratio of the normalized intensities of the resonance at δ 2.77, which corresponds to the bis-allylic protons of n-6 acyl chains (two protons) over the triplet at δ 0.97 that belongs to n-3 fatty acids and corresponds to three protons. The relationship is Cn-6/ Cn-3 = 3/2 IA/IB, where IA and IB are the integrals of the signals at δ 2.77 and δ 0.97, respectively. n-6 fatty acids are determined from the relationship Cn-6 = 3/2In-6/S, where In-6 is integral of the bis-allylic protons at δ 2.77.
Example 4, trans fatty acids: Trans fatty acids can be calculated from the equation Ctrans= Itrans/S, where Itrans is the integral of the signal at δ 0.91. The present sample contained 3.07% of trans fatty acids, as determined by 1H NMR using the 850 MHz instrument. The same sample analyzed in a 500 MHz instrument was found to contain 3.03% of trans fatty acids.
Example 5, saturated fatty acids (SFA): The concentration of SFA can be calculated from the equation CSFA = S–Cn-3 –Cn-6 –Cn-9 –Cn-1 – Ctrans. n-9 fatty acids (mainly oleic acid) can be quantified according to the equation Cn-9 = (3/4Q – 3/2In-6)/S, where Q is the integral of the allylic protons of n-6 and n-9 at δ 2.01. The amount of SFA in a commercially available fish oil sample was found to be 36.1%. The same sample analyzed with 13C NMR was found to contain 33.8% SFA. SFA represent a group of various FA (e.g. stearic and palmitic) with different MW and thus their concentration if fish oil cannot be expressed in mg/g.
Example 6, total sterols: The amount of total sterols (free and esterified) can be determined by the signal of the methyl protons at carbon 18 which appears at δ 0.68, using the equation C = Iste/S. The molar ratio of total sterols in a commercially available fish oil sample was found to be 0.32%. BHT can also be used for the determination of the absolute concentration of sterols. The main sterols in fish oil are cholesterol and vitamin D (or its precursor 7-dehydrocholesterol) and are often added in the supplements. These compounds have a very similar MW. Therefore, results can be expressed in mg/g and are calculated according to the equation C = 2/3 ISTE/IISA × MWSTE/m, where MWSTE is the molecular mass (386) of cholesterol, which constitutes the majority of the sterolic fraction in fish oil20. The amount of sterols in the same sample using BHT was 3.8 mg/g of fish oil. The individual determination of cholesterol (δ 0.680) and 7-dehydrocholesterol (δ 0.678) is feasible on an 850 MHz instrument after the application of a window function for resolution enhancement.
13C NMR Analysis
Figure 2 illustrates the 13C NMR spectra acquired on (A) an 850 MHz and (B) a 500 MHz instrument in the carbonyl carbon area. The two spectra are very similar and can provide the same amount of information. The 13C NMR spectrum can be successfully used for the analysis of additional fatty acids such as stearidonic (SDA) and eicosatetraenoic (ETA) acids, however more scans are required for samples in which these acids are in lower concentrations. The 13C spectra are characterized by high resolution because of the large spectral width and the application of broadband decoupling, which eliminates the effect of scalar coupling and produces singlets. For this reason, there is limited overlapping even when using a 500 MHz instrument.
The 13C NMR spectrum is much more informative compared to the 1H NMR spectrum and can provide more comprehensive quantitative data because less signal overlapping is observed (Figures 1 and 2). The most useful spectral region of the 13C spectrum is the carbonyl carbon region because it provides quantitative information for a large number of fatty acids as well as for their positional distribution on the glycerol skeleton19,21,22. The methyl group area from δ 14.5 to δ 13.5 can be used for the quick determination of the total amount of n-3, n-6, n-9 and saturated fatty acids (SFA), as well as trans fatty acids. However, in the 500 MHz NMR spectrometer, there is a partial overlapping of the n-6 and n-9 saturated fatty acids (SFA). The application of a window function for resolution enhancement may solve this problem although the 850 MHz instrument is still considered a more reliable option. The olefinic region of the carbon spectrum can be used for the total amount of n-3 and n-1 acyl chains as well as for determination of individual fatty acids such as DHA, EPA, Arachidonic acid (AA), Linolenic (Ln) n-3, and oleic acid (OL) (see Table 2). 13C NMR can be also applied for the characterization of fish oil from other sources, such as supplements rich in ethyl esters (EE) using the carbon signals at δ 14.31 (methyl) and δ 60.20 (methylene).
For carbon analysis, fatty acids can be determined by dividing the integral of the appropriate aliphatic, olefinic, and carbonyl signals with the total integral of all acyl chains, according to the general relationship C = I/S (2), where C is the concentration of the analyte in mole (%), I is the integral of a resonance that is uniquely attributed to the lipid of interest, and S is the total integral of signal(s) that represents the total lipid content of the sample. The total integral S of acyl chains can be determined by integrating the region from δ 175 to δ 171 and is set to 100.
Quantification of fatty acids in mg/g of fish oil is performed using an internal standard on the basis of the following relationship: C = I/IIS × A× MW/m (3), where C is the concentration of the analyte in mg/g of fish oil, I is the integral of a resonance that is uniquely attributed to the lipid of interest, IIS is the area of a carbon signal that belongs uniquely to the internal standard, A is the millimoles of internal standard, MW is the molecular weight of the compound of interest (for fatty acids expressed in methyl esters), and m is the amount of fish oil in g. The 13C-NMR signals of fish oil fatty acids that can be used for quantitation purposes are shown in Table 2, whereas the complete NMR assignment of the 13C NMR spectrum can be found elsewhere19.
Example 1, EPA at sn-2 position: The amount (%) of EPA on the sn-2 position is calculated by dividing the integral of the signal at δ 172.56 by S. The amount of EPA at the sn-2 position in a commercially available sample was found to be 3.4% using the 850 MHz instrument. Using the same spectrometer and BHT as an internal standard, the amount of EPA at the sn-2 position expressed in mg/g of fish oil is 29.73 mg/g. The same sample analyzed in a 500 MHz instrument was found to contain 3.6% or 31.39 mg/g of EPA in the sn-2 position. Similar results can be obtained when calculating the relative molecular ratios of EPA at sn-2 using a fully decoupled spectrum. This is because the carbonyl carbon of EPA is affected by proton decoupling to the same negligible degree as the other carbonyl carbons, which are used as reference. However, large deviations are observed when using BHT, because the carbon of BHT at δ 151.45, which is used for quantification, receive a different NOE enhancement compared to the carbonyl carbons of fatty acids. For that reason, the fully decoupled spectrum should be avoided when using internal standards or integrating carbons with different multiplicities.
Example 2, total amount of DHA: The total amount (%) of DHA is simply calculated by adding the amounts of DHA in sn-1,3 and sn-2 position as determined by the NMR signals at δ 172.48 and δ 172.08, respectively. The same sample analyzed with 1H NMR (see example 1 of 1H analysis) was found to contain 10.3% DHA according to 13C NMR analysis. The amount of DHA can also be expressed in mg/g by using an internal standard and Equation 3. The total amount of DHA was 103.25 mg/g.
Example 3, total amount of SDA: The total amount (%) of SDA is determined by adding the integrals of the signals at δ 172.99 and δ 172.60 which belong to the carbonyl carbons of SDA on position sn-1,3 and sn-2, respectively, then dividing the sum by S. The sample analyzed was found to contain 3.93% SDA or 34.54 mg/g.
Example 4, n-3 Ln: n-3 Ln (%) can be determined by dividing the integral of the signal at δ 131.85 with the integral S. The molar ratio of n-3 Ln in the analyzed fish oil sample was 0.7%. The absolute concentration using BHT was calculated as 5.5 mg/g.
Example 5, trans fatty acids: The molar ratio of trans fatty acids is determined by dividing the integral of the signal at δ 13.80 with S. The analysis of the same sample that was analyzed with 1H NMR and was found to be 3.07% of trans FA, was also analyzed with 13C NMR and its trans fatty acid content was found to be 3.42%. The 13C NMR analysis of the same sample on a 500 MHz instrument showed a 3.64% content of trans fatty acids. The amount of trans FA in mmol/g of fish oil can be determined using BHT as an internal standard and the equation C = I/IIS × A/m, however results cannot be expressed in mg/g because the peak at δ 13.80 corresponds to various trans fatty acids, mainly trans DHA and trans EPA, with different MW.
Example 6, EE: The concentration of EE in a fish oil sample is calculated by dividing the integral of the spectral area from δ 60.50 to δ 60.00, which corresponds to the methylene carbons of the EE of various fatty acids, with S. The analysis of an EE fish oil sample showed that it consisted of 100% EE. It should be noted that in EE samples, EPA can be calculated either by the carbonyl peak at δ 173.60 or by the methylene EE carbon at δ 60.20, whereas DHA can be calculated using the signal at δ 60.31 and/or the signal at δ 173.09.
A complete list of the diagnostic signals that can be used for quantification purposes with 13C and 1H NMR analysis can be found in Tables 1 and 2, respectively, whereas a detailed description of the equations that can be used for this analysis can be found elsewhere19.
NMR can additionally be applied for the assessment of the oxidation status of fish oil supplements. Figure 3 compares the 1H NMR spectra of a fish oil sample under two oxidation conditions; exposure to heating and exposure to ultraviolet (UV) light. Lipid oxidation is a complicated process, and the composition of oxidation products depends on the conditions of oxidation. The main oxidation products are hydroperoxides (δ 8.0-8.8), conjugated dienes hydroperoxides (δ 5.4-6.7), and aldehydes (δ 9.0- 10).
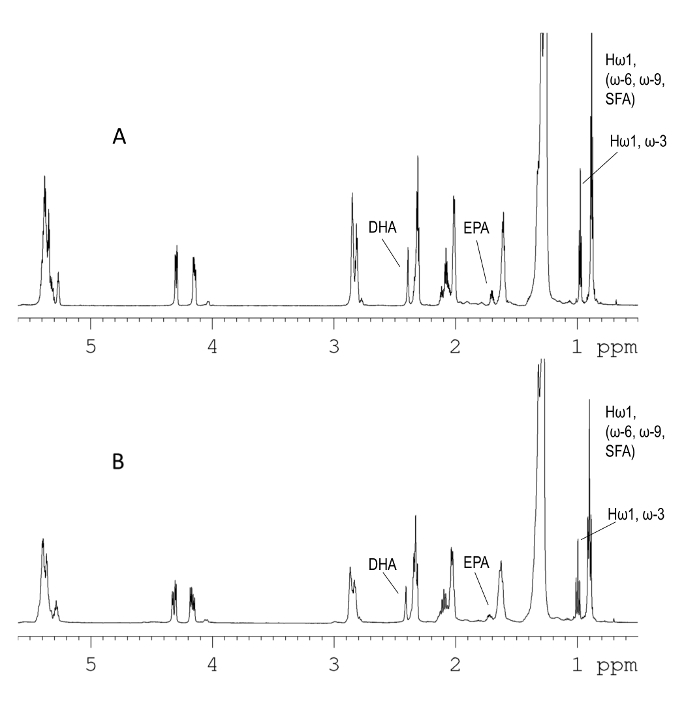
Figure 1. The 1H NMR analysis. 850.23 (A) and 500.20 MHz (B) 1H-NMR spectrum of a fish oil supplement in CDCl3 solution. The NMR signals of EPA and DHA that can be used for their determination are shown. The peak at δ 0.97 can be used for the determination of the total amount of n-3 fatty acids. The envelop at δ 1.39-1.20 is cropped, as it belongs to the methylene protons of all fatty chains and cannot be used for any identification or quantification purposes. The 1H NMR spectrum is characterized by a narrower spectral width (SW) compared to the 13C NMR spectrum and thus by lower spectral resolution. Please click here to view a larger version of this figure.
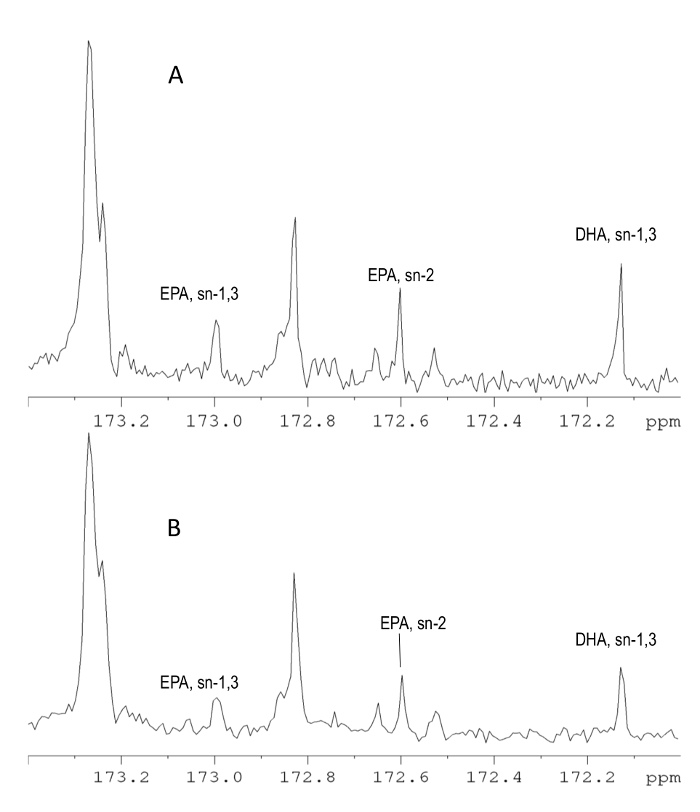
Figure 2. The 13C NMR analysis. 213.81 (A) and 125.77 MHz (B) 13C-NMR spectrum of a fish oil supplement in CDCl3 solution in the carbonyl carbon region. The NMR signals of EPA and DHA on sn-1,3 and sn-2 position are shown. These signals can be used for the quantitative determination of EPA and DHA. Although the spectra recorded at 213.81 MHz are characterized by a higher resolution and sensitivity, the 125.77 MHz spectra can also be used for the determination of the major compounds. The application of decoupling in the 13C NMR experiment eliminates the effect of scalar coupling between the carbon and hydrogen nuclei and thus the signals appear as singlets making the analysis easier compared to the 1H NMR spectrum. Please click here to view a larger version of this figure.
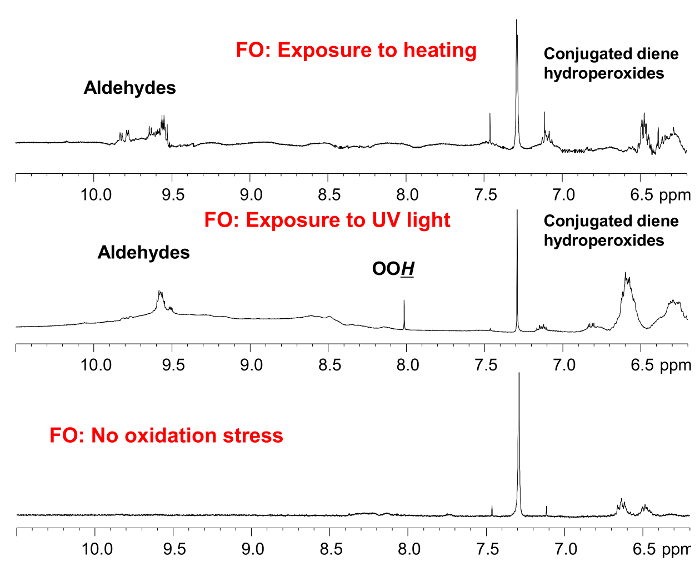
Figure 3. Fish oil oxidation. The 1H NMR spectrum of oxidized fish oil depends on the oxidation conditions. The resonances attributed to hydroperoxides (δ 8.0-8.8), conjugated dienes hydroperoxides (δ 5.4-6.7), and aldehydes are shown. Please click here to view a larger version of this figure.
| δ ppm | Proton | Compound |
| 0.677 | CH3 (18) | Cholesterol |
| 0.678 | CH3 (18) | 7-dehydrocholesterol |
| 0.88 | CH2CH3 (t), Jω1, ω2 = 7.27 Hz | n-9, SFA acyl chains |
| 0.883 | CH2CH3 (t), Jω1, ω2 = 7.08 Hz | n-6 acyl chains |
| 0.911 | CH2CH3 (t), Jω1, ω2 = 7.65 Hz | Trans acyl chains |
| 0.973 | CH2CH3 (t), Jω1, ω2 = 7.63 Hz | n-3 acyl chains |
| 1.25 | CH2CH3 (t), J = 7.20 Hz | Ethyl esters |
| 1.697 | OCOCH2CH2 (t), JHα, Ηβ = Hz | EPA acyl chain |
| 2.391 | OCOCH2CH2 (t) | DHA acyl chain |
| 2.772 | CH=CHCH2CH=CH | n-6 acyl chains |
| 2.81 | CH=CHCH2CH=CH | n-3 acyl chains |
| 3.593 | 3’a-CH2OCO | Glycerol of 1-MAG |
| 3.722 | 3’a, 3’b-CH2OCO (br) | Glycerol of 1,2-DAG |
| 4.073 | 2’-CHOH (br) | Glycerol of 1,3-DAG |
| 4.121 | CH2CH3 multiplet | Ethyl esters |
| 4.173 | 1’b, 3’b-CH2OCO (dd) | Glycerol of 1,3-DAG |
| 4.238 | 1’a-CH2OCO (dd) | Glycerol of 1,2-DAG |
| 4.329 | 1’b-CH2OCO (dd) | Glycerol of 1,2-DAG |
| 4.989 | -CH=CH2 cis (dd) | n-1 acyl chains |
| 5.052 | -CH=CH2 trans (dd) | n-1 acyl chains |
| 5.082 | 2’-CHOCO | Glycerol of 1,2-DAG |
| 5.268 | 2’-CHOCO | Glycerol of TAG |
| 5.436 | CH=CHCH2CH=CH2 | n-1 acyl chains |
| 5.818 | -CH=CH2 | n-1 acyl chains |
Table 1: The assignment of the 1H NMR spectrum. The 1H-NMR chemical shifts of fish oil fatty acid signals that can be used for quantification purposes in CDCl3 solution are presented. The chemical shifts are measured in ppm and provide information about the chemical environment of the nuclei.
| δ ppm | Carbon |
| 173.24 | C1 SFA (sn-1,3) |
| 172.21 | C1 OL, LO (sn-1,3) |
| 173.16 | C1 ETA (sn-1,3) |
| 173.13 | C1 DPA (sn-1,3) |
| 173.03 | C1 SDA (sn-1,3) |
| 172.97 | C1 EPA (sn-1,3) |
| 172.73 | C1 ETA (sn-2) |
| 172.69 | C1 DPA (sn-2) |
| 172.61 | C1 SDA (sn-2) |
| 172.56 | C1 EPA (sn-2) |
| 172.48 | C1 DHA (sn-1,3) |
| 172.08 | C1 DHA (sn-2) |
| 136.8 | Cω1, n-1 |
| 131.85 | Cω3 LN |
| 130.37 | C15 AA |
| 130.11 | C9 LN |
| 130.06 | C13 LO |
| 129.54 | C5 DHA sn-2 |
| 129.47 | C5 DHA sn-1,3 |
| 128.94 | C5 EPA |
| 128.76 | C6 EPA |
| 128.45 | C17 n-3 |
| 127.71 | n-3 |
| 127.53 | C4 DHA sn-2 |
| 127.5 | C4 DHA sn-1,3 |
| 126.86 | Cω4, all n-3 |
| 114.71 | Cω2, n-1 |
| 60.08 | DHA, Ethyl esters |
| 59.96 | EPA, Ethyl esters |
| 59.95-59.85 | Other FA, Ethyl esters |
| 33.48 | C2 EPA sn-2 |
| 33.32 | C2 EPA sn-1,3 |
| 31.44 | C3 n-1 |
| 27.05 | Allylic n-6 |
| 26.49 | C4 EPA sn-1,3 |
| 26.47 | C4 EPA sn-2 |
| 24.6 | C3 EPA |
| 24.48 | C3 SDA sn-1,3 |
| 24.44 | C3 SDA sn-2 |
| 14.27 | Cω1, all n-3 |
| 14.13 | Cω1, SFA |
| 14.11 | Cω1, OL |
| 14.07 | Cω1, LO |
| 13.8 | Cω1, trans FA |
Table 2: The assignment of the 13C NMR spectrum. The 13C-NMR chemical shifts of fish oil fatty acid signals that can be used for quantitation purposes in CDCl3 solution are presented.
Supplemental Figure S1: Comparison between the 13C NMR spectra acquired using the standard broadband decoupling (A) and the inverse gated decoupling (B) pulse sequences. The spectra were recorded for the same sample with the same number of scans, processed with the same processing parameters and are shown with the same scale factor. Please click here to download this figure.
