The following protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of New England. The following protocol is for preparing glial cultures from 4 adult mouse spinal cords.
1. Preparation of Solutions in a Culture Hood under Aseptic Conditions
- Prepare culture media: complete Dulbecco's modification of Eagle's media (cDMEM) containing DMEM (with 4.5 g/L glucose), 10% Fetal Bovine Serum (FBS), 2 mM L-glutamine, 100 IU/mL penicillin, 100 mg/mL streptomycin, 250 ng/mL Amphotericin B, and 50 µM 2-mercaptoethanol (2-ME; see 1.1.1). Mix and filter sterilize all other components and then add FBS. Store the culture media at 4 oC.
- Prepare 50 mM 2-ME/phosphate-buffered saline (PBS) stock solution: add 100 µL of concentrated 2-ME (14.3 M) into 28.6 mL of 1x PBS, and filter sterilize (the solution can be stored at 4 oC for several months). Use 1 mL of 2-ME stock solution for every 1,000 mL of culture media.
- Preparation of the density gradient media
- Prepare a stock isotonic density gradient media solution (e.g., 100% Percoll) from 1 part 10x regular sterile PBS and 9 parts sterile stock density gradient media (e.g., Percoll).
- Dilute the 100% density gradient media (made in 1.2.1) with regular 1x sterile PBS to make a 20% density gradient media (e.g., 10 mL of 100% density gradient media with 40 mL of 1x PBS) and store it at 4 oC. Bring the 20% density gradient media to room temperature (RT) before preparing the cell culture.
- Preparation of solutions from the papain dissociation system
NOTE: The papain dissociation system can be identified in the Materials Table. Other similar dissociation kits (including those assembled in-house) can also be used. All components must be sterile.- Add 32 mL of Earle's Balanced Salt Solution (EBSS) to the ovomucoid inhibitor mixture (powder).
- Add 5 mL of EBSS to one papain vial (powder). Place the papain vial in a 37 oC water bath for 10 min or until the papain is dissolved.
- Add 500 µL of EBSS to one DNase vial (powder). Mix gently.
- Add 250 µL of the DNase solution (above) to the papain vial.
- Calculate the total amount of the above papain/DNase mixture needed (approximately 800 µL of papain/DNase mixture per mouse spinal cord) and transfer the needed mixture into a 50 mL tube for tissue digestion (step 3.2). Store the leftover in the original vial at 4 °C for at least two weeks.
- Keep all components from the kits on ice until needed.
- Prepare spinal cord collection tubes: one sterile 15 mL tube with 5 mL of Hank's balanced Salt Solution (HBSS; from the papain dissociation system, for collecting spinal cords) and one sterile 15 mL tube with 1x PBS (for filling up the 5 mL syringe, see step 2.4). In addition, prepare one sterile petri dish (35 mm or 60 mm) with 5 mL of HBSS and keep it in the culture hood.
2. Spinal Cord Collection
NOTE: All equipment must be sterile. Perform the collection in a designated area approved by the IACUC.
- Obtain the following tools for harvesting mouse spinal cords: straight sharp/blunt 18-cm surgical scissors, a 12-cm standard scalpel (#3 solid), carbon steel sterile scalpel blades #10, a small animal decapitator, 12-cm standard curved forceps, and a sterile 20G needle attached to a 5-mL syringe.
- Euthanize the animal with CO2 and disinfect the mouse head and the back region with 70% ethanol (EtOH).
- Decapitate animal with the decapitator. Make a middle longitudinal incision on the lower back to expose the muscle layer. Make a clean transection through the vertebrate column at the hip level (also cutting through the muscle layer that covers the vertebrate column) with the sterile straight surgical scissors (18 cm).
- Place the animal on a sheet of fresh disposable wipe. Carefully insert the 20 G needle (attached to a 5 mL syringe filled with sterile 1x PBS (step 1.4)) into the spinal column towards the rostral side. Hold the animal down tightly and push the syringe quickly. The whole spinal cord should come out from the cervical end. Collect the spinal cord into the 15 mL tube containing HBSS.
- Repeat step 2.4 for the rest of the mice. Between each animal, disinfect all used instruments with 70% EtOH.
NOTE: Typically, a single 12-well plate can be established for every 4 mouse spinal cords (see 4.3).
3. Preparation of the Single Cell Suspension
Note: Perform steps 3 and 4 in a culture hood to keep everything sterile.
- Transfer all spinal cords into the HBSS-containing petri dish (step 1.4). Cut each of the spinal cords into many fine, small pieces with sterile scissors and forceps. Transfer the spinal cord tissue to the 50-mL conical tube containing the prepared papain/DNase enzyme mixture (step 1.3.5) using sterile forceps or a 10 mL pipette (avoid adding HBSS to the enzyme mixture, as this will further dilute the prepared enzyme solution and may result in decreased performance of the enzyme).
- Vortex the tube gently to mix. Incubate the tube at 37 oC for 1 h in an incubator/shaker with orbital shaking at 150 rpm.
- Vortex the tube again and vigorously triturate the enzyme solution with the tissue using a 5 mL pipette to promote further dissociation.
- Transfer the cell suspension into a 15-mL tube and centrifuge at 300 x g for 5 min at RT.
- During centrifugation, mix 2.7 mL EBSS with 300 µL of reconstituted albumin-ovomucoid inhibitor solution (1.3.1) in a sterile tube. Add 150 µL of the DNase solution (step 1.3.3).
- Following centrifugation, remove the supernatant and resuspend the cell pellet with the solution prepared above (step 3.5). Vortex well to break the cell pellet.
- Add 3 mL of reconstituted albumin-ovomucoid inhibitor solution (step 1.4.1) to the cell suspension. Centrifuge cells at 70 x g for 6 min at RT. Remove the supernatant (which contains membrane fragments).
4. Further Removal of Myelin from the Single Cell Suspension
- Add 8 mL of 20% density gradient media (prepared in step 1.2.2) into the tube containing the cell pellet, vortex gently to disrupt the pellet, and centrifuge the cells at 800 x g for 30 min at RT without braking. Carefully remove the top layer of debris (mostly myelin) and the supernatant, but keep the pellet.
- To remove remnants of the density gradient, wash the cells by resuspending the cell pellet with 8 mL of a diluted cDMEM (1 part cDMEM and 2 parts HBSS). Centrifuge the cells at 400 x g for 10 min at 4 oC. Remove the supernatant and wash the cells again with the diluted cDMEM (above) in the same manner. Keep the cells on ice until seeding them.
- Remove the supernatant and resuspend the cell pellet in culture media (cDMEM supplied with 2-ME (prepared in step 1.1)). For one 12-well plate, use 3 mL x 4 (number of mice used) + 2 mL = 14 mL media. This will ensure that there is sufficient cell suspension for the entire plate (12 wells) and will provide for extra wells that can be used to determine the average cell number per well and the microglial content of the culture. If other types of culture vessels will be used, calculate the total volume of needed culture media proportionally.
- Add 1 mL of the cell suspension into each well of a 12-well plate.
- Incubate the cells at 35.9 oC with 5% CO2.
- Change the media (remove old media via aspiration) on D 1 and then every 3 – 4 d thereafter (typically change the media on D: 1, 4, 8, and 11, and then use the cells on day 12).
NOTE: On day 1, the culture may contain significant amounts of debris due to the residue myelin from the spinal cord tissue; thus, changing the media on day 1 is recommended (please see the Discussion for more information). Cultures are ready for treatment between D 12 – 14. Cells usually are 80% confluent at D 12 and can be near 100% confluent by D 14. Typically, on D 12, there are about 100,000 cells per well in a 12-well plate.

Figure 2: Representative images of mixed glial cells at different times after the establishment of the mixed glia culture. Primary spinal cord mixed glia were prepared from adult C57Bl/6 mice. Representative images show the progress of the glial culture after plating. At day 1, some cells are attached to the culture plate, but they are still mostly round. There are also many floating cells and significant debris. At day 4, the majority of the cells are attached to the culture plate. Cells appear ramified with visible processes. Cells are sporadically distributed and cultures are about 20-30% confluent at this time. At day 8, cultures are between 50-60% confluent. Some areas of the cultures have large patches of cells. Other areas of the cultures have sparse growth of cells. Some cells within the patches take on a “square-like” appearance. At day 12, cultures are at or above 80% confluent (about 90% confluent in the image shown here). Cell are in the log phase of growth at this time and between days 12-14 is the optimal time for using the glia cells for experiments. Please click here to view a larger version of this figure.
- Examine the microglial content using cells from the extra wells (step 4.3) via standard fluorescence-activated cell sorting (FACS) staining protocol11 using the combination of anti-mouse-CD45 and anti-mouse CD11b monoclonal antibodies. CD45+CD11b+ cells are identified as microglia.
This method can be used to prepare mixed glial cells from both mice and rats. The average total cell number per well in a 12-well plate on D 12 post-initiation of the culture should be relatively stable, with around 100,000 cells per well when cells are derived from mouse spinal cords. Glial cells obtained from this method can be used in experiments that are designed to examine adult spinal cord glial responses upon administration of substances and agents of interest. Figure 3 provides an example of cytokine and chemokine responses from one typical experiment in which adult spinal cord microglia were obtained from adult BALB/c mice and treated with LPS (Salmonella Minnesota Re595) on D 13 post-initiation of the culture. Supernatants were collected 24 h post-LPS treatment for the determination of cytokine and chemokine levels via an enzyme-linked immunosorbent assay (ELISA) using commercially-available kits.
When this adult glial culture system was first established, cells were incubated in a standard culture environment at 37 oC. Under this condition, the average microglial content ranged from 5 – 10%. However, it was observed that despite the use of consistent culture techniques, cultures would in some instances have either very low microglial content (<2%) or relatively high microglial content (15 - 20%)10. It can be frustrating to obtain cultures that have very few microglia when the experimental results rely on the microglial responses within the mixed culture. Following the suggestion of Dr. Alejandro M. S. Mayer (Department of Pharmacology, Chicago College of Osteopathic Medicine)12, the method was modified by growing the mixed glial cells at 35.9 oC. This resulted in a more consistent and higher microglial yield (ranging between 10 – 40% and remaining mostly around 20%) within the majority of cultures. This improvement is illustrated in Figure 4. As reported previously, microglia are estimated to make up 5 – 21% of the CNS glial population in adult mice13-15. Although both culture conditions provide cultures that contain similar amounts of microglia as estimated in vivo, the modified culture condition (35.9 oC) is more appropriate for experiments in which it is critical to examine the responses from both astrocytes and microglia.

Figure 1: Calcitonin Gene-related Peptide (CGRP)-induced Chemokine Production by Mixed Glial Cells. Mixed glial cells prepared from neonatal BALB/c mouse brains (left) or adult BALB/c mouse spinal cords (right) were treated with various doses of CGRP. Levels of several chemokines in culture supernatants were determined via multiplex assay (performed by the manufacturer) at optimal times (mean ± SEM, n = 2 – 6). Please click here to view a larger version of this figure.
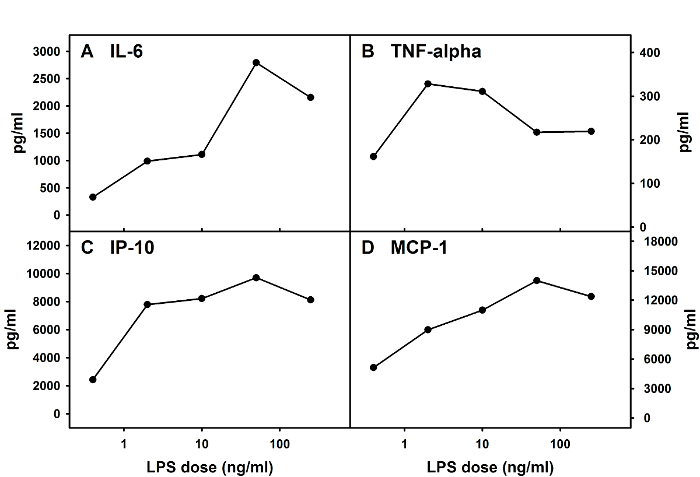
Figure 3: Cytokine and Chemokine Responses of Mouse Adult Spinal Cord Mixed Glia upon LPS Stimulation. Adult spinal cord mixed glial cells were prepared from BALB/c mice and stimulated with various doses of LPS. Levels of IL-6 (A), tumor necrosis factor (TNF)-alpha (B), interferon-gamma-inducible protein 10 (IP-10, also known as CXCL10) (C), and monocyte chemoattractant protein 1 (MCP-1, also known as CCL2) (D) in culture supernatants were determined via ELISAs at 24 h post-LPS treatment. Please click here to view a larger version of this figure.
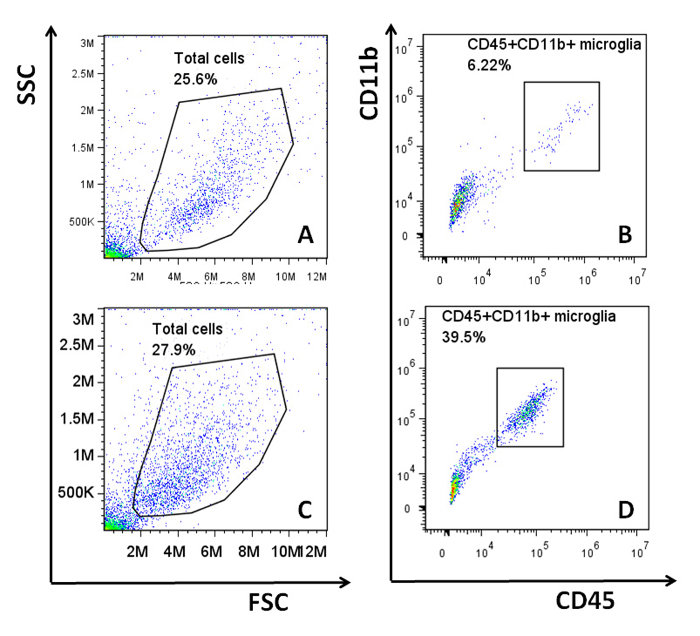
Figure 4: Microglial Content in Adult Mixed Glial Cultures. Mixed glial cells were generated from adult C57B6/L mice and cultured at either 37 oC or 35.9 oC. Microglial content from each culture was analyzed via flow cytometry using APC-anti-mouse-CD45 (clone 30-F11) and FITC-anti-mouse-CD11b (clone M1/70). Representative plots from the flow cytometric analyses are shown. The total cell populations from the cultures were identified in plots A (37 oC) and C (35.9 oC), and the microglial populations (CD45+CD11b+ cells) were further isolated from the respective total cell populations in B (37 oC) and D (35.9 oC). Flow cytometric analysis was performed, and all data were analyzed as previously described5. Please click here to view a larger version of this figure.
