The N-triphosPh ligand (1) and the ruthenium complex series: Ru(CO)2{N(CH2PPh2)3}-κ3P] (2), [Ru(CO3)(CO){N(CH2PPh2)3}-κ3P] (3) and [Ru(H)2(CO){N(CH2PPh2)3}-κ3P] (4) were characterized via 1H, 13C{1H}, 30P{1H} NMR spectroscopy, FT-IR, ESI mass spectrometry and elemental analysis. Representative 1H and 30P{1H} NMR data are shown in Table 1. In the case of complexes 2, 3 and 4 single crystal X-ray analysis unequivocally confirms their molecular structures. 30P{1H} NMR spectroscopy is a particularly useful technique for studying these complexes as characteristic shifts to higher frequency relative to free ligand and splitting patterns can be used to identify successful ligand coordination and identify particular geometries of complexes.
The free ligand N-triphosPh (1) displays a single resonance in the 30P{1H} NMR spectrum (CDCl3, 162 MHz) at -28.9 ppm. Occasionally, oxide peaks may appear at higher frequencies in the 30P{1H} NMR spectrum if due care is not take to exclude oxygen during the reaction or when making a solution for NMR spectroscopy. Reaction of N-triphosPh (1) with [Ru3(CO)12] results in the dicarbonyl complex [Ru(CO)2{N(CH2PPh2)3}-κ3P] (2) that shows a characteristic higher frequency shift of a singlet to 8.3 ppm in the 30P{1H} NMR spectrum (C6D6, 162 MHz), indicating that all the phosphine arms are coordinated to the Ru center and are in the same chemical environment. The X-ray crystal structure also confirmed this (Figure 4A).
Oxidation of 2 to gives the ruthenium(II) carbonate complex [Ru(CO3)(CO){N(CH2PPh2)3}-κ3P] (3), simply by bubbling molecular oxygen through a suspension of 2 in toluene. A significant change in the 30P{1H} NMR spectrum is seen compared to 2. A characteristic triplet and doublet, AB2 coupling pattern, in seen in the 30P{1H} NMR spectrum of 3 with resonances at –23.5 ppm (triplet) and 15.9 ppm (doublet) as there are now two different phosphorus environments, a result of the loss of symmetry on the formation of a carbonate. FT-IR can be used to confirm characteristic κ2-carbonate stretches at 1,565 and 1,434 cm-1. Single crystal X-ray diffraction analysis also confirmed this structure (Figure 4B).
Hydrogenation of 3 under 15 bar hydrogen pressure gives the dihydrogen complex [Ru(H)2(CO){N(CH2PPh2)3}-κ3P] (4) (Figure 2). The 30P{1H} NMR spectrum in C6D6 gave a doublet at 8.5 ppm and triplet at 18.8 ppm, indicating two different phosphorus environments. The 1H NMR spectrum shows characteristic hydride resonances in the low frequency region of the spectrum as a multiplet centered around –6.50 ppm. Single crystal X-ray diffraction analysis also confirmed the structure of dihydride complex (Figure 4C).
Reaction of 4 with NH4PF6 in acetonitrile results in the loss of a hydride ligand and the formation of molecular H2, and [RuH(CO)(MeCN){N(CH2PPh2)3}-κ3P](PF6) (5) (Figure 3). The 30P{1H} NMR spectrum is further complicated as there are now three different phosphorus environments owing to the three different trans ligands coordinating to the ruthenium center. A multiplet and two doublet-of-doublets at –12.4, 3.9 ppm and 26.5 ppm are seen (Figure 5). In the low frequency region of the 1H NMR spectrum a pseudo doublet-of-triplets for 5 is seen at –6.3 ppm (Figure 6). The addition of levulinic acid to 5 gives the complex [Ru(CO){N(CH2PPh2)3}-κ3P{CH3CO(CH2)2CO2H}-κ2O](PF6) (6) (Figure 3). The 1H NMR spectrum of 6 after 21 hr shows the complete disappearance of the Ru–H signal (Figure 5) and 30P{1H} NMR spectrum shows a pseudo triplet at –16.2 ppm and doublet 19.8 ppm (Figure 6).
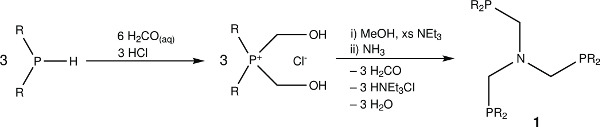
Figure 1. The chemical structures of the triphosphine ligand N-triphosPh and its generation synthetic scheme.
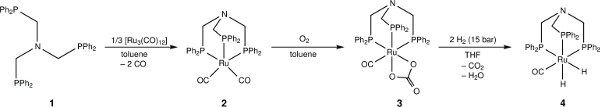
Figure 2. The chemical structure of ruthenium complexes of N-triphosPh and a synthetic scheme for their sequential preparation.
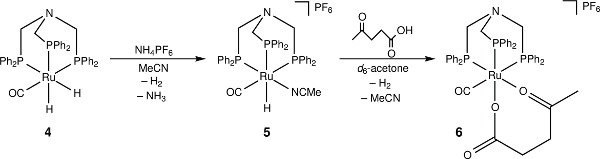
Figure 3. The activation of [RuH2(CO){N(CH2PPh2)3}-κ3P] with NH4PF6 and subsequent coordination with levulinic acid.
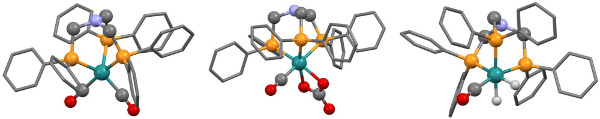
Figure 4. X-ray crystal structures of three Ru–N-triphosPh complexes, bearing (A) dicarbonyl (complex 2) (B) carbonate carbonyl (complex 3) and (C) dihydride (complex 4) ancillary ligands. These structures were obtained by Andrew J. P. White of Imperial College London. Note, crystals of [Ru(CO3)(CO){N(CH2PPh2)3}-κ3P] were found to contain two crystallographically independent complexes, only one of which is shown here. Please click here to view a larger version of this figure.
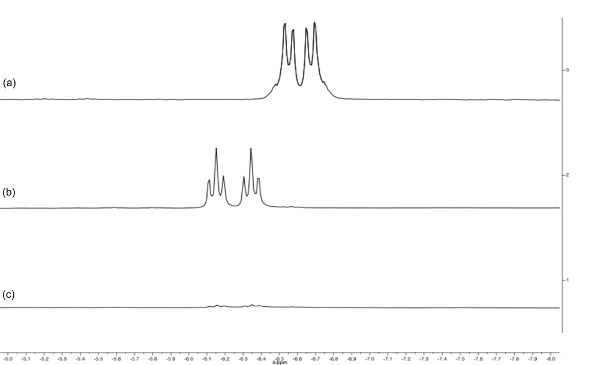
Figure 5. Stacked spectra of the hydride region (–5 to –8 ppm) of the 1H NMR spectra of [RuH2(CO){N(CH2PPh2)3}-κ3P] (a, d8-toluene, 400 MHz), [RuH(CO)(MeCN){N(CH2PPh2)3}-κ3P]PF6 (b, d6-acetone, 400 MHz) and [Ru(CO){N(CH2PPh2)3}-κ3P{CH3CO(CH2)2CO2H}-κ2O](PF6) (c, d6-acetone, 400 MHz). Note the change as the complex is converted from a dihydride (pseudo doublet-of-doublets) to a monohydride (doublet-of-triplets) and finally to complete loss of hydride ligands.
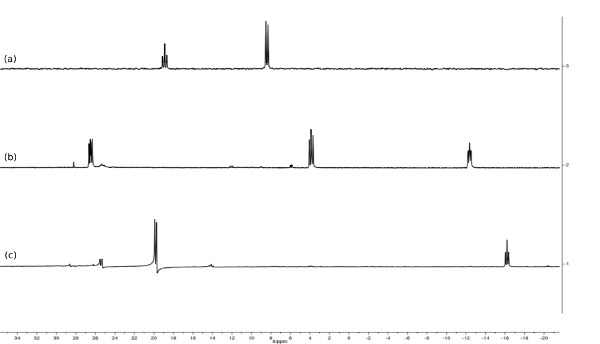
Figure 6. Stacked 30P{1H} spectra of [RuH2(CO){N(CH2PPh2)3}-κ3P] (a, d8-toluene, 162 MHz), [RuH(CO)(MeCN){N(CH2PPh2)3}-κ3P]PF6 (b, d6-acetone, 162 MHz) and [Ru(CO){N(CH2PPh2)3}-κ3P{CH3CO(CH2)2CO2H}-κ2O](PF6) (c, d6-acetone, 162 MHz). Note how the splitting pattern and number of resonances changes with identity of the ancillary ligands.
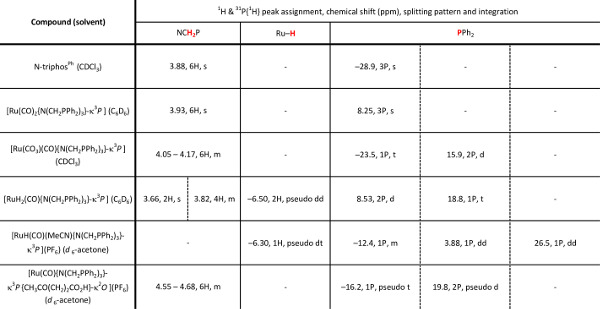
Table 1. The 1H and 30P{1H} NMR characterization data of the triphosphine ligand and subsequent ruthenium complexes. d = doublet, t = triplet, m = multiplet; pseudo splitting patterns are observed when two separate resonances have very similar chemical shifts and coupling constants.
