Metal-free Synthesis of Ynones from Acyl Chlorides and Potassium Alkynyltrifluoroborate Salts
Summary
The goal of this manuscript is to demonstrate a straightforward method for the preparation of ynones from acyl chloride and potassium alkynyltrifluoroborate salt starting materials. The one-pot reaction proceeds rapidly in the presence of boron trichloride without exclusion of air and moisture.
Abstract
Ynones are a valuable functional group and building block in organic synthesis. Ynones serve as a precursor to many important organic functional groups and scaffolds. Traditional methods for the preparation of ynones are associated with drawbacks including harsh conditions, multiple purification steps, and the presence of unwanted byproducts. An alternative method for the straightforward preparation of ynones from acyl chlorides and potassium alkynyltrifluoroborate salts is described herein. The adoption of organotrifluoroborate salts as an alternative to organometallic reagents for the formation of new carbon-carbon bonds has a number of advantages. Potassium organotrifluoroborate salts are shelf stable, have good functional group tolerance, low toxicity, and a wide variety are straightforward to prepare. The title reaction proceeds rapidly at ambient temperature in the presence of a Lewis acid without the exclusion of air and moisture. Fair to excellent yields may be obtained via reaction of various aryl and alkyl acid chlorides with alkynyltrifluoroborate salts in the presence of boron trichloride.
Introduction
The intention of this video is to demonstrate a straightforward approach for the preparation of compounds containing an ynone functional group from convenient starting materials. Ynones are valuable building blocks in organic chemistry that have been shown to have biomedical and material significance. Ynones are precursors to valuable organic functional groups including pyrimidines,1,2 quinolones,3 furans,4 pyrazoles,5,6 flavones,7 oximes,8 and chiral propargylic alcohols.9-11 A more convenient method for the preparation of ynones has been sought as a result of the drawbacks of traditional methods including poor functional group tolerance and tedious synthetic routes.
The reaction of metal12,13 and metalloid14 acetylides with acyl chlorides is one common route for the preparation of ynones. Alternatively, the synthesis of ynones from acyl chloride can be achieved via two-step procedures using Weinreb amides and organolithium or Grignard reagents.15 Another prevalent approach includes the addition of organolithium or Grignard reagents to an aldehyde, which is followed by the oxidation of a secondary alcohol to the corresponding ketone.16-19 Poor functional group tolerance of metal acetylides and the need to purify synthetic intermediates after each step are main deficiencies of the aforementioned methods. Transition-metal-catalyzed carbonylative couplings have recently emerged as an alternative approach for the preparation of ynones.20-22 Unfortunately, in addition to cost and toxicity associated with transition metals, metal-catalyzed carbonylative reactions often require elevated CO pressures and suffer from the presence of an undesired Sonogashira coupling byproduct. Given their utility in organic synthesis as well as the drawbacks associated with traditional synthetic methods, the development of a more straightforward method for the preparation of ynones is appealing.
Potassium organotrifluoroborate salts have recently emerged as versatile reagents in organic synthesis. Advantages including ease of preparation,23 inherent stability, low toxicity, and good functional group tolerance have made organotrifluoroborate salts attractive synthetic reagents.24-27 Organotrifluoroborate salts have been used primarily as a bench-stable equivalent of boronic acids for palladium-catalyzed Suzuki-Miyaura coupling.26 Recently, following a seminal work by Matteson and co-workers,28 Bode, Molander and others have highlighted the utility of organotrifluoroborates as reagents in non-metal catalyzed reactions.29-33 The field of transition-metal-free reactions of trifluoroborates is still in its infancy stage. Given the great potential for use of organotrifluoroborate salts in non-metal catalyzed organic synthesis, we sought to develop a novel method for the preparation of ynones from acyl chlorides and alkynyltrifluoroborate salts.
Protocol
1. Synthesis of Ynone
- Using an analytical balance, weigh 0.15 mmol of desired potassium alkynyltrifluoroborate salt (1.5 equivalents) and add to a clean vial fitted with a magnetic stir bar.
- Using a glass plunger syringe fitted with a needle, add 0.5 ml of anhydrous DCM to the vial to afford a 0.3 M solution of trifluoroborate.
NOTE: Since this is a new methodology, the reactions have been performed for proof of concept. In order to reduce waste they have been performed on a small scale. The reaction may be scaled to produce larger yields as required. - Using a glass syringe, add 0.15 mmol of 1.0 M boron trichloride solution (1.5 equivalents, 150 µl) dropwise to the reaction vessel at ambient temperature (20 °C) with stirring. Replace cap. Hold or clamp the capped reaction vessel such that the reaction solution is below the water line in the sonicator. Sonicate the solution for 30 sec at 20 °C and a frequency of 40 kHz.
- Stir the reaction solution at room temperature (20 °C) for an additional 20 min. Depending on the nature of the alkynyltrifluoroborate starting material, observe a color change. A color change from white to tan or dark brown, red, or purple is common.
- Add 0.1 mmol of the desired acyl chloride (1.0 equivalent) to the vial at ambient temperature (20 °C) with stirring. Alternatively, transfer the contents of the reaction vessel to a flask charged with a magnetic stir bar and 0.1 mmol of acyl chloride. Replace cap. Stir the reaction solution at ambient temperature (20 °C) for 30 min.
NOTE: Generally, a change to a very dark color is an indication that the reaction is taking place. - Using a thin layer chromatography (TLC)34 monitor the progress of the reaction by comparing with appropriate acyl chloride standard.
NOTE: Depending on the nature of the starting materials, the reaction may not go to completion. The acyl chloride starting material has been observed as a higher running spot than the final product for all of the ynones prepared to date using this method.- Prepare the acyl chloride standard by diluting approximately 10 mg of the acyl chloride starting material with 1 ml of ethyl acetate in a small vial.
- Using a TLC spotter, spot the reaction solution and the standard on the starting line of the TLC plate. A co-spot with both the reaction solution and standard may be useful to determine if there is still starting material present in the reaction solution.
- Determine a suitable TLC mobile phase by trial and error. For the preparation of product 1a, a mobile phase consisting of 1:30 ethyl acetate:hexanes results in Rf values of approximately 0.5 and 0.8 for the ynone product and benzoyl chloride starting material, respectively.
- Visualize the developed TLC plate under UV light. Use a pencil to mark any visible spots.
2. Aqueous Workup
- Quench the reaction by adding a 1 ml of cold water (approximately 5 °C) to the reaction vessel. Using a Pasteur pipette, transfer the solution to a clean separatory funnel. Further dilute the contents of the separatory funnel with 10 ml cold water and extract the product once into 15 ml ethyl acetate.
- Discard the bottom aqueous layer and wash the organic layer one additional time with 10 ml of water and one time with 10 ml of brine. Collect the organic layer into a clean Erlenmeyer flask and dry with magnesium sulfate. Filter the solution into a clean round bottom flask using gravity filtration.
3. Purification of Ynone
- Using thin layer chromatography, determine a suitable mobile phase for flash chromatography purification of the product. A retention factor (Rf) of approximately 0.5 is generally sufficient to separate the product from any remaining starting material and side products.
NOTE: In the case of poor separation, a less polar solvent system may be used such that the product has a lower Rf value. Depending on the polarity of the starting materials, a hexanes:ethyl acetate mixture in the range of 30:1 to 5:1 is generally suitable. - Prepare a column containing silica wet with the appropriate mobile phase. Add a layer of sand to the top of the column in order to avoid disturbing silica upon addition of the product and eluent.
- Evaporate the workup solution using a rotary evaporator with the bath temperature at 40 °C and the rotation set to 120 rpm. Dissolve the contents of the flask in a 2 ml of mobile phase and carefully load the solution evenly onto the column. Ensure that the silica is wet but the solvent line does not exceed the height of the silica. Allow some solvent to elute such that the product is on the silica. Add a small amount of mobile phase and repeat.
- Fill the column with the mobile phase determined in step 3.1; in the case of product 1a, a 30:1 hexanes:ethyl acetate mixture is appropriate. Collect the fractions in test tubes using flash chromatography to speed up the purification process. Simply connect an adaptor with tubing to an air outlet. Adjust the air pressure such that the eluent flows through the column at a suitable rate.
- Spot each of the column fractions on a TLC plate. Optionally, spot the acyl chloride standard as a reference. Develop the TLC using the mobile phase deemed appropriate in step 1.8. Mark any spots visible under UV light, noting that the product is expected to have a lower Rf than the acyl chloride standard.
- Collect all fractions containing visible spots into a large round bottom flask. If more than one spot with a lower Rf than the starting material is present, collect them in separate flasks. Evaporate the solvent using a rotary evaporator and dry the material collected under high vacuum for two hours.
- If required, perform a pentane wash on a small column in order to further purify an ynone product prepared from an electron poor acyl chloride starting material. On a small scale, this may be performed using a Pasteur pipette as the column.
- Dissolve the product in a minimum of chloroform, DCM, or diethyl ether and load onto a silica column wet with pentane. Elute 1-2 column lengths of pentane through the column and collect into a waste beaker.
- Elute the pure ynone product from the pentane column using diethyl ether or ethyl acetate. Collect eluent in a separate flask, evaporate, and place under high vacuum for a minimum of two hours. Obtain approximately 10 mg of product for characterization by NMR spectroscopy.
4. Characterization of Ynone
- Characterize and assess the purity of the final product using 1H and 13C NMR spectroscopy.34 Typically, the chemical shift of the carbonyl carbon appears near 177 ppm on the 13C NMR spectrum. The two SP carbons of the ynone functional group are represented by characteristic peaks near 87 and 93 ppm.
- Use infrared spectroscopy34 to identify the characteristic carbonyl peak of the ynone product. Electron delocalization from the adjacent triple bond usually causes the carbonyl peak to appear between 1,600 and 1,650 cm-1, depending on the electronic characteristics of the compound.
- Determine the molecular mass of the product and further validate the identity using high resolution mass spectrometry.34
Representative Results
Initial efforts were focused around the preparation of ynone 1a from phenylacetylene trifluoroborate S1 and benzoyl chloride (Figure 1). Table 1 illustrates the optimization steps performed including screening of various Lewis acids, solvents, as well as examination of the effect of water on the reaction. Next, the scope of the reaction has been explored by submitting phenylacetylene trifluoroborate to the optimized conditions in the presence of a variety of acyl chlorides (Figure 2). Modest to excellent yields may be obtained depending on the nature of the acyl chloride substrate. The scope of the reaction has further been evaluated through the preparation of several additional examples of alkynyltrifluoroborate salts. Figure 3 illustrates selected examples of other alkynyltrifluoroborate salts that can be employed in the preparation of ynones under the developed conditions. Derivatives of phenylacetylene trifluoroborate bearing electron donating substituents afforded the corresponding ynone products in good to excellent yields while aliphatic derivatives of alkynyltrifluoroborate salts proved to be slightly less reactive affording modest yields.
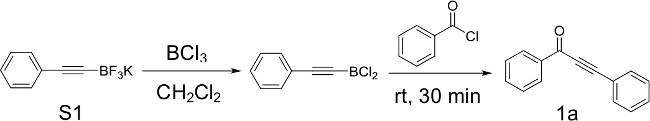
Figure 1: Preparation of Ynone 1a from phenylacetylene trifluoroborate S1 and benzoyl chloride. Scheme illustrating the conditions for the preparation of ynone 1a including formation of the reactive dichloroborane intermediate.

Figure 2: Representative 1H NMR spectrum of ynone 1a. Chemical shifts and relative integrations of characteristic protons are labeled.
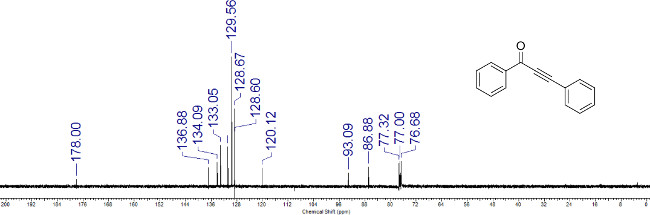
Figure 3: Representative 13C NMR spectrum of ynone 1a. Chemical shifts of characteristic carbons are labeled.
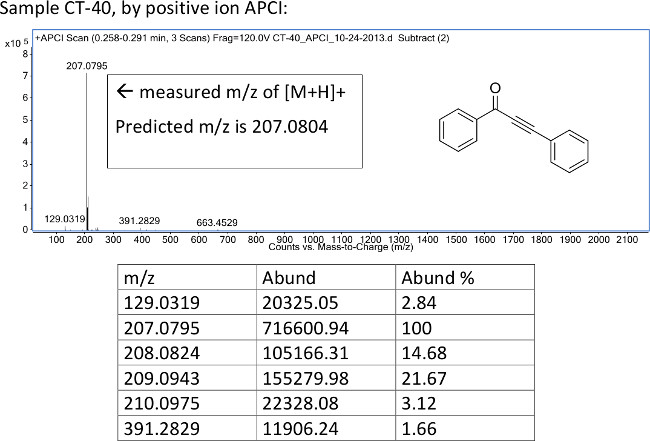
Figure 4: Representative HRMS spectrum of ynone 1a obtained via atmospheric-pressure chemical ionization. The measured m/z value of the [M+H]+ ion is reported.
Figure 5: Reactions of phenylacetylenetrifluoroborate salt S1 with acyl chlorides. Structures and yields for products produced by varying the identity of the acyl chloride starting material are illustrated. Reactions were run with 1 equiv of acyl chloride, 1.5 equiv potassium phenylacetylenetrifluoroborate S1, and 1.5 equiv boron trichloride.
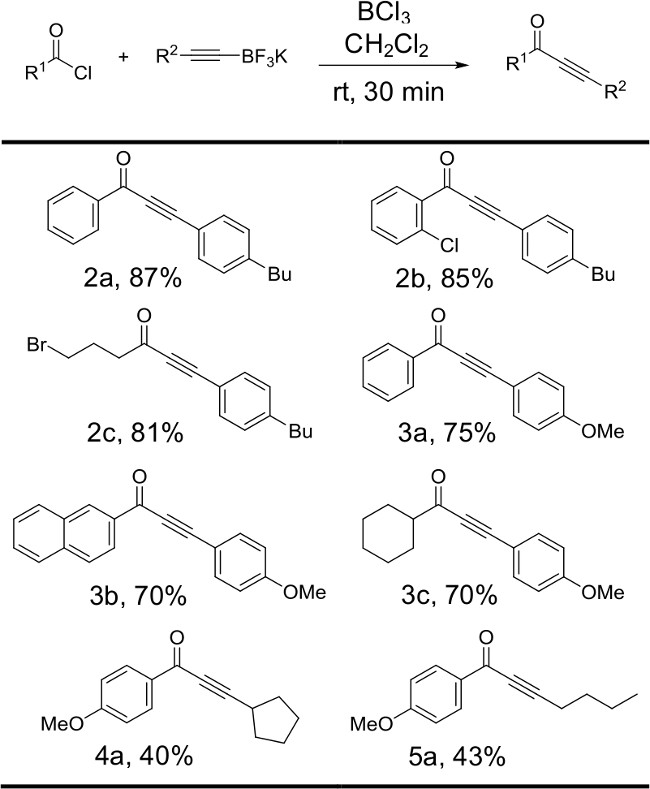
Figure 6: Reactions of various alkynyltrifluoroborates with acyl chlorides. Structures and yields for products produced by varying the identity of the alkynyltrifluoroborate starting material are illustrated. Reactions were run with 1 equiv of acyl chloride, 2.5 equiv potassium organotrifluoroborate salt and 2.5 equiv boron trichloride.
| Entry | Lewis acid | Lewis acid (equiv) | Conditions | Yield (%) |
| 1b | SiO2 | 16.5 | CH2Cl2 | 0 |
| 2 | SiCl4 | 1 | CH2Cl2 | trace |
| 3 | BF3·OEt2 | 1 | CH2Cl2 | trace |
| 4b | FeCl3 | 1 | CH2Cl2 | 24 |
| 5c | AlCl3 | 1 | CH2Cl2 | 62 |
| 6d | AlCl3 | 1 | CH2Cl2 | 66 |
| 7e | AlCl3 | 1 | CH2Cl2 | 60 |
| 8 | AlCl3 | 1 | THF | trace |
| 9 | AlCl3 | 1 | toluene | trace |
| 10 | AlCl3 | 2 | ClCH2CH2Cl | 33 |
| 11 | AlCl3 | 1 | DMSO | trace |
| 12 | AlCl3 | 2 | acetonitrile | trace |
| 13 | AlCl3·6H2O | 1 | CH2Cl2 | 0 |
| 14b | BCl3 | 1.5 | CH2Cl2 | 67 |
| 15 | BBr3 | 1 | CH2Cl2 | 20 |
Table 1: Optimization of conditions for the preparation of ynone 1aa. Yield of product 1a upon variation of reaction conditions and Lewis acid catalyst. aReactions were run with 1 equiv benzoyl chloride and 1 equiv potassium phenylacetylenetrifluoroborate S1. b1.5 equiv S1. cAnhydrous conditions. dThe reaction was done in non-dried glassware under air. eAnhydrous conditions + 1 µl of water.
Discussion
Table 1 illustrates the steps taken to optimize the conditions for the reaction of phenylacetylene trifluoroborate with benzoyl chloride to form the corresponding ynone product. Initially, catalysts known to convert organotrifluoroborates to organodifluoroboranes were tested. Unfortunately Silica gel,35 silicon tetrachloride,36 and boron trifluoride31,32 did not promote the formation of the desired ynone (Table 1, entries 1-3). The use of chlorinated Lewis acid catalysts proved to be more successful. A low yield of the desired ynone 1a was obtained in the presence of an iron(III) chloride catalyst (Table 1, entry 4). Next, aluminum(III) chloride was investigated as a result of its well-established ability to promote oxocarbenium ion formation in Friedel-Crafts acylations.37-39 The desired product was obtained in 62% yield when an aluminum(III) chloride catalyst was employed.
Further optimization revealed that air and moisture have little effect on the yield of the reaction (Table 1, entries 5-7). As a result, subsequent reactions were performed in non-dried glassware in the presence of air. Attempts to optimize the solvent revealed that dichloromethane (DCM) is particularly well suited to the reaction (Table 1, entries 8-12). Inconsistencies in the results of reactions catalyzed by aluminum(III) chloride prompted the exploration of alternative catalysts. Commercially available aluminum(III) chloride hexahydrate was completely inactive under the reaction conditions (Table 1, entry 13). This is a good indicator that the formation of aluminum(III) chloride hydrate inhibits the reaction. Boron trichloride was found to produce similar yields with better consistency (Table 1, entry 14).
Upon interaction of the potassium alkynyltrifluoroborate with boron trichloride, a more reactive organodichloroborane species is formed.40 This initial step is critical for the reaction with the acyl chloride and formation of the ynone to proceed. Since organotrifluoroborate salts are not soluble in DCM, the reaction takes place as a heterogeneous mixture. After addition of boron trichloride, the solution is sonicated order to facilitate formation of the reactive dichloroborane species by increasing the surface area of the trifluoroborate salt available to react. Application of ultrasound waves to the reaction mixture causes mechanical effects through the generation of cavitation bubbles. During sonication, collapse of cavitation bubbles in the fluid results in localized areas of high temperatures and pressures.41 Shock waves are produced that create microscopic turbulence resulting in an increase in kinetic energy of the solid trifluoroborate salts. The increase in energy of the system during sonication promotes fragmentation of the trifluoroborate salt resulting increased surface area available to interact with boron trichloride. Sonication of the reaction mixture prior to addition of the acyl chloride starting material ensures the efficient formation of the reactive alkynyldichloroborane species without the need for more forcing conditions or longer reaction times.
Figure 2 illustrates the results obtained when phenylacetylene trifluoroborate was reacted with a variety of acyl chlorides under the optimized reaction conditions. Neutral aromatic (1b, 1c) and aliphatic (1j-l) acyl chlorides furnish the corresponding ynones in synthetically useful yields. Those acyl chlorides bearing electron donating groups (1d-g) provide excellent yields while electron withdrawing groups result in comparatively modest yields (1h, 1i, 1m). Interestingly, when the electron withdrawing group is located in the ortho position (1i, 59%), a significant yield increase is observed in comparison to the analogous para substituted acyl chloride (1h, 30%). The steric interaction of the substituent in the ortho- position may force the carbonyl functional group out of the plane, thereby offsetting the electron-withdrawing character of the aromatic ring. It is worth noting that 4-bromobutyryl chloride reacted to afford the desired product 1m in 39% yield. To our knowledge, this is the first protocol for the synthesis of ynones to tolerate an alkyl bromide functional group. Occasionally, when the acyl chloride starting material is neutral or electron deficient, aliphatic impurities appear on the proton NMR. This may necessitate a pentane wash in order to further purify the product. While possible, it is not economical to perform the pentane wash during the first purification stage since pentane is costly in comparison to hexanes. Completing the secondary purification separately on a smaller scale such as a Pasteur pipette column significantly reduces the amount of pentane required.
Figure 3 illustrates the effect of the identity of the alkynyltrifluoroborate salt on the yield of the reaction. In general, derivatives of the phenylacetylene trifluoroborate salt bearing electron-donating substituents on the aromatic ring reacted with aromatic and aliphatic acyl chlorides to produce the desired ynones in good to excellent yields (2a-c, 3a-c). Aliphatic alkynyltrifluoroborate salts proved to be less reactive substrates. Modest yields have been obtained when hexynyl- and cyclopentylethynyltrifluoroborate salts were reacted with electron-rich benzoyl chloride derivatives (4a, 5a).
In conclusion, a novel method for the preparation of ynones from acyl chlorides and potassium alkynyltrifluoroborate salts has been developed. The yields obtained for the synthesis of ynones by this method range from modest to excellent depending on the nature of the acyl chloride and trifluoroborate starting materials. In general those starting materials bearing electron donating substituents undergo the reaction more readily than starting materials bearing neutral and electron withdrawing functional groups. The value of this approach lies in the operational simplicity and functional group tolerance of the method. This straightforward, one-pot reaction proceeds rapidly at ambient temperature in the presence of boron trichloride without exclusion of air and moisture. This convenient method may be employed in the preparation of ynones in modest to excellent yields from a variety of acyl chlorides and alkynyltrifluoroborate salts.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the UOIT start-up fund. We thank Mr. Matthew Baxter (UOIT) for his assistance in the laboratory.
Materials
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
| Sonicator | VGT | 1860QT | Ultrasonic cleaner. Similar sonication devices may be used. |
| Dichloromethane | Sigma | 270997 | Anhydrous |
| Boron trichloride solution | Sigma | 178934 | 1M solution in DCM |
| Acyl chloride | Sigma | Various | Acyl chlorides from other suppliers such as Alfa Aesar may be used. Caution – refer to MSDS for safety information. |
| Potassium alkynyltrifluoroborate salt | N/A | N/A | Synthesized23 from terminal alkyne |
| Ethyl acetate | ACP | E-2000 | ACS grade |
| Hexanes | ACP | H-3500 | ACS grade |
| Pentane | Sigma | 236705 | Anhydrous |
| Magnesium sulfate | Sigma | M7506 | |
| Filter paper | Whatman | 1093 126 | Student grade. This speceific variety is not necessary. |
| Silica Gel 60 | EMD | 1.11567.9026 | Particle size 0.040-0.063 |
References
- Bannwarth, P., Valleix, A., Grée, D., Grée, R. Flexible Synthesis of Pyrimidines with Chiral Monofluorinated and Difluoromethyl Side Chains. J. Org. Chem. 74, 4646-4649 (2009).
- Karpov, A. S., Merkul, E., Rominger, F., Müller, T. J. Concise Syntheses of Meridianins by Carbonylative Alkynylation and a Four‐Component Pyrimidine Synthesis. Angew. Chem. Int. Ed. 44, 6951-6956 (2005).
- Arcadi, A., Aschi, M., Marinelli, F., Verdecchia, M. Pd-catalyzed regioselective hydroarylation of α-(2-aminoaryl)-α, β-ynones with organoboron derivatives as a tool for the synthesis of quinolines: experimental evidence and quantum-chemical calculations. Tetrahedron. 64, 5354-5361 (2008).
- Lee, C. G., Lee, K. Y., Lee, S., Kim, J. N. Chemical transformation of Baylis–Hillman adducts: the reaction of methyl 3-arylamino-2-methylene-3-phenylpropanoates in polyphosphoric acid. Tetrahedron. 61, 1493-1499 (2005).
- Kirkham, J. D., Edeson, S. J., Stokes, S., Harrity, J. P. Synthesis of Ynone Trifluoroborates toward Functionalized Pyrazoles. Org. Lett. 14, 5354-5357 (2012).
- Mohamed Ahmed, M. S., Kobayashi, K., Mori, A. One-pot construction of pyrazoles and isoxazoles with palladium-catalyzed four-component coupling. Org. Lett. 7, 4487-4489 (2005).
- Awuah, E., Capretta, A. Access to Flavones via a Microwave-Assisted, One-Pot Sonogashira− Carbonylation− Annulation Reaction. Org. Lett. 11, 3210-3213 (2009).
- She, Z., et al. Synthesis of Trisubstituted Isoxazoles by Palladium (II)-Catalyzed Cascade Cyclization–Alkenylation of 2-Alkyn-1-one O-Methyl Oximes. J. Org. Chem. 77, 3627-3633 (2012).
- Corey, E., Helal, C. J. Novel electronic effects of remote substituents on the oxazaborolidine-catalyzed enantioselective reduction of ketones. Tetrahedron Lett. 36, 9153-9156 (1995).
- Matsumura, K., Hashiguchi, S., Ikariya, T., Noyori, R. Asymmetric transfer hydrogenation of α, β-acetylenic ketones. J. Am. Chem. Soc. 119, 8738-8739 (1997).
- Midland, M. M., McDowell, D. C., Hatch, R. L., Tramontano, A. Reduction of. alpha.,. beta.-acetylenic ketones with B-3-pinanyl-9-borabicyclo [3.3. 1] nonane. High asymmetric induction in aliphatic systems. J. Am. Chem. Soc. 102, 867-869 (1980).
- Logue, M. W., Moore, G. L. Cuprous trimethylsilylacetylide. Preparation and reaction with acid chlorides. J. Org. Chem. 40, 131-132 (1975).
- Normant, J. Organocopper (I) Compounds and Organocuprates in Synthesis. Synthesis. , 63-80 (1972).
- Logue, M. W., Teng, K. Palladium-catalyzed reactions of acyl chlorides with (1-alkynyl) tributylstannanes. A convenient synthesis for 1-alkynyl ketones. J. Org. Chem. 47, 2549-2553 (1982).
- Nahm, S., Weinreb, S. M. N-Methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 22, 3815-3818 (1981).
- Baba, T., Kizuka, H., Handa, H., Ono, Y. Reaction of ketones or aldehydes with 1-alkynes over solid-base catalysts. Appl. Catal., A. 194, 203-211 (2000).
- Brown, H. C., Garg, C. P. A simple procedure for the chromic acid oxidation of alcohols to ketones of high purity. J. Am. Chem. Soc. 83, 2952-2953 (1961).
- Nishimura, T., Onoue, T., Ohe, K., Uemura, S. Palladium (II)-catalyzed oxidation of alcohols to aldehydes and ketones by molecular oxygen. J. Org. Chem. 64, 6750-6755 (1999).
- Pu, L. Asymmetric alkynylzinc additions to aldehydes and ketones. Tetrahedron. 59, 9873-9886 (2003).
- Kim, W., Park, K., Park, A., Choe, J., Lee, S. Pd-Catalyzed Selective Carbonylative and Non-carbonylative Couplings of Propiolic Acid: One-Pot Synthesis of Diarylalkynones. Org. Lett. 15, 1654-1657 (2013).
- Kobayashi, T., Tanaka, M., Sakakura, T. Palladium(ii) Complex-catalysed Formation of a-Keto Acids via Double Carbonylation of Organic. Halides J. Chem. Soc., Chem. Commun. , 837-838 (1985).
- Mohamed Ahmed, M. S., Mori, A. Carbonylative Sonogashira coupling of terminal alkynes with aqueous ammonia. Org. Lett. 5, 3057-3060 (2003).
- Vieira, A. S., et al. Nucleophilic Addition of Potassium Alkynyltrifluoroborates to d-Glucal Mediated by BF3· OEt2: Highly Stereoselective Synthesis of α-C-glycosides. Org. Lett. 10, 5215-5218 (2008).
- Darses, S., Genet, J. -. P. Potassium organotrifluoroborates: new perspectives in organic synthesis. Chem. Rev. 108, 288-325 (2008).
- Darses, S., Genet, J. P. Potassium trifluoro (organo) borates: new perspectives in organic chemistry. Eur. J. Org. Chem. 2003, 4313-4327 (2003).
- Molander, G. A., Ellis, N. Organotrifluoroborates: Protected Boronic Acids That Expand the Versatility of the Suzuki Coupling Reaction. Acc. Chem. Res. 40, 275-286 (2007).
- Stefani, H. A., Cella, R., Vieira, A. S. Recent advances in organotrifluoroborates chemistry. Tetrahedron. 63, 3623-3658 (2007).
- Matteson, D. S., Kim, G. Y. Asymmetric alkyldifluoroboranes and their use in secondary amine synthesis. Org. Lett. 4, 2153-2155 (2002).
- Dumas, A. M., Bode, J. W. . Synthesis of Acyltrifluoroborates. Org. Lett. 14, 2138-2141 (2012).
- Dumas, A. M., Molander, G. A., Bode, J. W. Amide‐Forming Ligation of Acyltrifluoroborates and Hydroxylamines in Water. Angew. Chem. Int. Ed. 51, 5683-5686 (2012).
- Mitchell, T. A., Bode, J. W. Synthesis of Dialkyl Ethers from Organotrifluoroborates and Acetals. J. Am. Chem. Soc. 131, 18057-18059 (2009).
- Vo, C. -. V. T., Mitchell, T. A., Bode, J. W. Expanded substrate scope and improved reactivity of ether-forming cross-coupling reactions of organotrifluoroborates and acetals. J. Am. Chem. Soc. 133, 14082-14089 (2011).
- Zeng, J., Vedachalam, S., Xiang, S., Liu, X. -. W. Direct C-Glycosylation of Organotrifluoroborates with Glycosyl Fluorides and Its Application to the Total Synthesis of (+)-Varitriol. Org. Lett. 13, 42-45 (2010).
- Lehman, J. W. The student’s lab companion: laboratory techniques for organic chemistry: standard scale and microscale. Pearson College Div. , (2008).
- Molander, G. A., Cavalcanti, L. N., Canturk, B., Pan, P. -. S., Kennedy, L. E. Efficient Hydrolysis of Organotrifluoroborates via Silica Gel and Water. 74, 7364-7369 (2009).
- Molander, G. A., Ellis, N. Organotrifluoroborates: protected boronic acids that expand the versatility of the Suzuki coupling reaction. Acc. Chem. Res. 40, 275-286 (2007).
- Calloway, N. The Friedel-Crafts Syntheses. Chem. Rev. 17, 327-392 (1935).
- Gore, P. The Friedel-Crafts acylation reaction and its application to polycyclic aromatic hydrocarbons. Chem. Rev. 55, 229-281 (1955).
- Groves, J. The Friedel–Crafts acylation of alkenes. Chem. Soc. Rev. 1, 73-97 (1972).
- Kim, B. J., Matteson, D. S. Conversion of alkyltrifluoroborates into alkyldichloroboranes with tetrachlorosilane in coordinating solvents. Angew. Chem. 116, 3118-3120 (2004).
- Thompson, L., Doraiswamy, L. Sonochemistry: science and engineering. Ind. Eng. Chem. Res. 38, 1215-1249 (1999).
