1) Preparation of E. coli DH5α Cell Lysate
- Inoculate a 2mL culture (LB media in a test-tube) with the E. coli DH5α strain directly from a glycerol stock and incubate at 37°C and 250 rpm for 8 h. Use autoclaved LB media (10 g/l Bacto-tryptone, 5 g/L yeast extract, 10 g/L NaCl, pH 7.5).
- Inoculate 250 mL LB media in a 1 L shaker flask with baffles with the 2 mL culture and incubate over night at 37°C and 166 rpm in an incubator with orbital shaker.
- Inoculate four 5 L shaker flasks with baffles each containing 2.5 L LB media with the 250 mL culture (50 mL for each flask) and incubate the cultures at 37°C and 166 rpm until an OD600 of 0.8 is reached.
- Harvest the cells by centrifugation at 4°C, 3000x g for 20 min. Perform further handling at 0-4°C or on ice.
- Re-suspend the harvested cell material in Milli-Q water, combine in one centrifuge bucket and centrifuge for a further 30 min at 6000x g and 4°C.
- Store the resulting 20 g cell material at -20°C or -78°C.
- Re-suspend the cells in 100 mL ice cold cell opening buffer (6.7 mM MES, 6.7 mM NaOAc, 6.7 mM HEPES, 1 mM EDTA, 10 mM β-mercaptoethanol, 200 mM NaCl, pH 7.5, 10% (w/v) glycerol; 0.2 mM PMSF) and sonicate three times for 1 min in four 25 mL portions on ice using a sonifier (e.g. SONOPULS HD 2070 from BANDELIN electronic GmbH & Co. KG, maximal amplitude, continuous output).
- Centrifuge the lysate over night at 2370x g and 2°C.
- Concentrate the supernatant to 14 mL by ultrafiltration (e.g. using iCON Concentrators, 7 mL/9K, from Pierce) at 2370x g and 2°C.
- Deplete the viscous concentrate from small molecules like SAM or SAH by gel filtration at 2°C (e.g. Zeba Desalt Spin Columns, 10 mL, from Pierce; four columns; storage buffer removed at 1000x g for 2 min; four times equilibration with 5 mL ice cold cell opening buffer and buffer removed at 1000x g for 2 min, respectively; 3.5 mL concentrate applied to each column; 45 min centrifugation at 24x g, then two times 2 min at 1000x g)
- Supplement the resulting 13 mL lysate with 13 mL ice cold glycerol and Roche mini protease inhibitor cocktail tablets, EDTA free. Mix and dissolve the tablets.
- Store the lysate at -20°C
- Determine the total protein concentration by the Bradford assay (21 mg/ml in the present case). Bradford reagent: 100 mg Coomassie Brilliant Blue G-250 in 50 mL 95% ethanol, add 100 mL 85% (w/v) phosphoric acid, dilute to 1 L when the dye has completely dissolved, and filter through Whatman #1 paper just before use.
2) Capture Assay (A), Competition Control (C), Pulldown (PD), Competition Control of Pulldown (CPD), and Combined Capture Assay plus Pulldown (A+PD)
- For the capture experiments, the SAH caproKit (caprotec bioanalytics GmbH) was used, which includes the SAH-CC, free SAH as competitor, streptavidin-coated magnetic beads with 1 μm diameter (Dynabeads MyOne Streptavidin C1, Invitrogen Dynal), 5X capture buffer (5X CB, containing 100 mM HEPES, 250 mM potassium acetate, 50 mM magnesium acetate and 50% glycerol), and 5X wash buffer (5X WB, containing 250 mM Tris HCl, pH 7.5, 5 mM EDTA, 5 M NaCl, 42.5 μM octyl-β-D-glucopyranoside).
- For several parallel experiments it is recommended to prepare a master mix of water, capture buffer and E. coli lysate and to perform reactions within different tubes of one 200 μL-PCR tube strip (recommended 0.2 mL Thermo-Strip, Thermo Scientific, AB-1114). Here, quantities for one reaction tube are given. Results for five different experiments will be presented, which are capture assay (A), competition control (C), pulldown (PD), competition control of pulldown (CPD), and combined capture assay plus pulldown (A+PD).
- For each reaction, prepare 1.5 mL 1X WB by adding 0.3 mL 5X WB to 1.2 mL of Milli-Q water.
- Prepare SAH-CC loaded streptavidin-coated magnetic beads (caproBeads) in 200 μL PCR tube strips. Therefore, mix 25 μL of 100 μM SAH-CC with 50 μL of 10 mg/ml streptavidin-coated magnetic beads for each aliquot, vigorously shake the resulting suspensions at room temperature for 2 min to allow binding of the biotin moiety of the SAH-CC to the streptavidin on the magnetic bead surface, and collect the beads using a strong magnet (e.g. in the caps of the PCR tube strips using the caproMagTM magnetic device, caprotec bioanalytics GmbH). Discard the supernatants, re-suspend the resulting caproBeads in 200 μL WB, magnetically collect the caproBeads (in the caps of the PCR tube strips), and discard the supernating WB. Close tubes to avoid drying of the beads.
- Prepare aliquots of E.coli DH5α whole cell lysate in new PCR tubes at 0-4°C using a master mix (see 2.2). For one reaction, supplement a volume of Milli-Q water for a final reaction mix volume of 100 μL with 20 μL 5X CB. Mix, add 0.26 mg E. coli lysate, and gently mix by inversion. Only for C and CPD, add 20 μL 10 mM SAH competitor solution and gently mix by inversion (add Milli-Q water instead of SAH solution to A, PD, and A+PD). Draw a 1 μL sample from A for further analysis (see below).
- Suspend the caproBeads in the respective lysate and incubate for 3 h at 4°C keeping the beads in suspension by rotation to allow reversible binding of SAH binding proteins to the SAH selectivity function of the SAH-CC.
- Place the suspensions A, C, and A+PD in the caproBoxTM (device for irradiating biochemical samples with UV light and simultaneously cooling, caprotec bioanalytics GmbH) and irradiate for a total time of 30 min in the closed tubes between 0-4°C to form a covalent crosslink between the reactivity function of the SAH-CC to the SAH binding proteins. Therefore, remove the suspensions after irradiation intervals of 2.5 min, respectively, from the caproBoxTM, cool in ice water for ~15 s (especially the lids), mix several times by inversion, very shortly (~2 s) centrifuge to remove suspension remaining in the lids, and place back into the caproBoxTM for the next irradiation interval.
- Add 20 μL 10 mM SAH solution to A, or 20 μL Milli-Q water to C, PD, CPD, and A+PD and incubate the suspension for 10 min at 4°C to displace, in A, SAH binding proteins not crosslinked to the SAH-CC. Keep the beads in suspension by rotation or by intermittent manual re-suspension.
- Collect the caproBeads from the suspensions using a strong magnet (e.g. the caproMagTM), discard the supernatants, and wash the beads six times – by re-suspension and collection – with 200 μL 1X WB and once with 200 μL Milli-Q water.
- The beads can be stored in Milli-Q water for several weeks at 4°C. Alternative protocols exist for further processing of the captured proteins and their identification (see discussion).
- Wash the beads three times with 200 μL 60% acetonitrile (ACN) and release the captured proteins from the beads by 10 min incubation at room temperature under vigorous shaking with 200 μL 60% ACN/0.2% trifluoroacetic acid (TFA) (prepare freshly). Use LC-MS-grade reagents and water.
- Magnetically collect the beads, separate and evaporate the supernatant to dryness using a centrifugal evaporator (e.g. MiVac DNA concentrator from GeneVac, Inc., UK). Discard the beads.
3) SDS-PAGE of Captured Proteins
- For SDS-PAGE, dissolve the captured proteins released from the magnetic beads (evaporated ACN/TFA solutions from step 2.12) in 20 μL SDS sample buffer (50 mM Tris HCl, 320 mM β-mercaptoethanol, 2.5% SDS, 0.05% bromophenol blue, 10% glycerol, pH 6.8). Mix the 1 μL sample drawn from A (see step 2.5) with 19 μL SDS sample buffer; use 5 μL of this solution for SDS-PAGE analysis (0.25% of assay). Heat the samples in SDS sample buffer 10 min to 95°C and let allow to cool to room temperature.
- Analyze by SDS-PAGE (generic setup: OLS® ProPage 4-20% Tris/glycine pre-cast gels; OLS omniPAGE mini gel electrophoresis system; SDS running buffer: 25 mM tris base, 200 mM glycine, 0.1% SDS, pH 8.3; run time 90 min at a constant voltage of 180 V under ice cooling of the SDS running buffer).
- Silver stain the gel using an MS compatible silver stain method (e.g. ProteoSilver Silver Stain Kit from Sigma). A representative result is shown in Figure 2.
4) In-gel Tryptic Digest of Proteins and Peptide Extraction from Gel Bands
- Wash the silver stained gel at least three times with 100 mL Milli-Q water for 10 min after the silver stain stop solution was removed.
- Cut out the gel bands (e.g. using a clean scalpel and transfer into a 0.5 mL Eppendorf tube) and store at -20°C or directly process. Wash the gel bands for 15 min, respectively, with 100 μL water, 100 μL 50% ethanol, 100 μL water, 100 μL 50% ethanol, and for 5 min with pure ethanol. Repeat this washing procedure once again.
- Re-hydrate the gel band with 10 μL in-gel digestion solution (12.5 ng/μL sequencing grade trypsin in 50 mM ammonium bicarbonate, prepare by adding 7.5 μL 0.5 μg/μL trypsin solution in 1 mM HCl to 292.5 μL 50 mM ammonium bicarbonate) for 45 min at 4°C. Remove the supernatant and replace by 20 μL 50 mM ammonium bicarbonate (without trypsin) followed by incubation at 37°C over night while shaking.
- Collect the supernatant. For peptide extraction, incubate the gel band with 20 μL 5% formic acid (FA) for 15 min while shaking, add 20 μL ACN and incubated for another 15 min while shaking. Combine the supernatant with the previous supernatant and repeat the peptide extraction procedure once again.
- Evaporate the combined three supernatants to dryness, dissolve in 10 μL 5% FA while shaking and applying ultrasound (ultrasound bath, e.g. Sonorex from Bandelin, Germany) and proceed with desalting (step 5.2 and further).
5) Tryptic Digest of Captured Proteins and Preparation of Peptides for LC-MS/MS
- Dissolve the captured proteins released from the magnetic beads (evaporated ACN/TFA solutions from step 2.12) in 10 μL 50 mM ammonium bicarbonate using an ultrasound bath and vortexing, add 1 μL 0.5 μg/μL trypsin in 1 mM HCl and incubate the solution at 37°C over night.
- Desalt the solution containing the tryptic peptides of the captured proteins using C18 material (e.g. 2-10 μL StageTips, 20 μL tip, Proxeon Biosystems A/S, Odense, Denmark, manufacturer’s procedure: pre-condition with 10 μL 50% methanol/5% FA, equilibrate with 10 μL 5% FA, load with peptides of captured proteins, wash with 10 μL 5% FA, elute twice with 10 μL 50% methanol/5% FA).
- Evaporate the desalted peptides in 50% methanol/5% FA to dryness, dissolve in 5.5 μL 0.1% FA while shaking and applying ultrasound (ultrasound bath) and analyze the sample by nanoLC-MS/MS.
6) NanoLC-MS/MS Analysis
- Transfer sample into sample plate and place plate into the nano-flow liquid chromatography (nanoLC) system (e.g. Easy-nLC liquid chromatography system; Proxeon Biosystems A/S, Denmark).
- Use 0.1% FA in water as mobile phase A and 0.1% FA in ACN as mobile phase B. Only use LC-MS grade solvents.
- Load 5 μL of the peptide solution directly onto a pre-column (e.g. nanoflow Biosphere C18, 5 μm, 120 Å, 20 x 0.1 mm; NanoSeparations, Netherlands) coupled to an analytical column (e.g. nanoflow Biosphere C18, 5 μm, 120 Å, 100 x 0.075 mm; NanoSeparations, Netherlands) using 5% ACN/0.1% FA.
- During LC, elute peptides during an 80 min linear gradient from 5% ACN/0.1% FA to 40% ACN/0.1% FA followed by an additional 2 min to 100% ACN/0.1% FA and remaining at 100% ACN/0.1% FA for another 8 min with a controlled flow rate of 400 nL/min.
- Perform mass spectrometric (MS) analysis of eluted peptides on a high accuracy state-of-the-art mass spectrometer (e.g. LTQ Orbitrap XL mass spectrometer; Thermo FisherScientific, Germany, equipped with a nanoelectrospray ion source for electrospray ionization (ESI); Proxeon Biosystems A/S, Denmark).
- Perform the mass spectrometric analysis in the data-dependent mode to automatically switch between orbitrap-MS (profile mode) and LTQ-MS/MS (centroid mode) acquisition.
- Control the mass spectrometer duty cycle by setting the injection time automatic gain control.
- Acquire survey full-scan MS spectra (from m/z 300 to 2000) in the orbitrap with resolution r = 60,000 at m/z 400 (after accumulation to a target value of 500,000 charges in the linear ion trap).
- Set the instrument to sequentially isolate the most intense ions (up to five, depending on signal intensity) for fragmentation in the linear ion trap using collision-induced dissociation (CID) at a target value of 10,000 charges. The resulting fragment ions are recorded in the LTQ.
- For accurate mass measurements in the MS mode, use the singly charged polydimethylcyclosiloxane background ion (Si(CH3)2O)6H+ (m/z 445.120025) generated during the electrospray process from ambient air as the lock mass for real time internal recalibration.
- Dynamically exclude target ions already mass-selected for CID for the duration of 60 s.
- Set charge state screening and rejection of ions for CID with unassigned charge.
- Further mass spectrometric settings are as follows: set spray voltage to 1.6 kV, set temperature of the heated transfer capillary to 200 °C, and normalized collision energy is 35% for MS2. The minimal signal required for MS2 is 500 counts. Apply an activation q = 0.25 and an activation time of 30 ms for MS2 acquisitions.
- To clean the LC system, perform one blank run between two consecutive CCMS measurements.
7) Peptide and Protein Identification via Automated Sequence Database Search
- Use a protein identification algorithm to analyze the MS/MS data (in the present case stored in raw files), e.g. SEQUEST implemented in BioworksBrowser 3.3.1 SP1 (Thermo FisherScientific, Germany) and X!Tandem (The Global Proteome Machine Organization; version 2007.01.01.1) implemented in the Scaffold 3 software (version Scaffold_3_00_03, Proteome Software Inc., USA).
- Perform automated database searching against the most recent UniProtKB/Swiss-Prot database release www.expasy.org of the organism investigated (database used for present study: Escherichia coli, strain K12, release 57-11).
- Use the following settings for automated database searching within SEQUEST: 5 ppm precursor tolerance, 1 amu fragment ion tolerance, and full trypsin specificity allowing for up to two missed cleavages. Allow as variable modifications phosphorylation at serine, threonine, and tyrosine; oxidation of methionines; deamidation at asparagines and glutamine; acetylation at lysine and serine; formylation at lysine; and methylation at arginine, lysine, serine, threonine, and asparagine. Do not use fixed modifications in the database search.
- Load srf or dta and out files generated by SEQUEST into the Scaffold 3, which performs probability assessment of peptide assignments and protein identifications by combining SEQUEST and X!Tandem database searches. Scaffold is useful for easily comparing and visualizing protein lists from several samples (in the present case A, C, PD, CPD, and A+PD).
- Set the parameters within the Scaffold 3 software to consider only peptides with ≥ 95% probability as specified by the Peptide Prophet algorithm (ref.13). Set protein identification probabilities for multiple peptide assignments to ≥ 95% according to the Protein Prophet algorithm13.For single peptide protein identifications, arbitrarily set protein probability to ≥ 50% and manually inspect the corresponding peptide MS/MS spectra. Proteins that comprise similar peptides and could not be differentiated based on MS/MS analysis alone are grouped by the software to satisfy the principles of parsimony. The estimated false discovery rate of peptide identifications can be determined using the reversed protein database approach and should be < 1%.
- Representative results of CCMS experiments are given in Tables 1, 2, and Supplementary Table S1 (mind that the protein database is not up-to-date for some proteins, e.g. PrmB (aka YfcB) or RsmH (aka MraW)), as well as in Figure 3.
8) Representative Results
Table 1:
| Protein | ORF | MW/kDa | Description | Substrate | A | C | PD | CPD | A+PD |
| Dcm | b1961 | 53.5 | DNA-cytosine MTase | DNA (m5C) | 1 | 0 | 0 | 0 | 1 |
| RlmI | b0967 | 44.4 | 23S rRNA m5C1962 MTase | rRNA (m5C) | 17 | 0 | 17 | 0 | 20 |
| RlmL | b0948 | 78.9 | 23S rRNA m2G2445 MTase | rRNA (m2G) | 12 | 0 | 0 | 0 | 10 |
| TrmB | b2960 | 27.3 | tRNA (guanine-N(7)-)-MTase | tRNA (m7G) | 11 | 0 | 0 | 0 | 13 |
| CmoA | b1870 | 27.8 | tRNA (cmo5U34)-MTase | tRNA (mcmo5U) | 7 | 0 | 0 | 0 | 4 |
| RsmG | b3740 | 23.4 | 16S rRNA m7G MTase | rRNA (m7G) | 6 | 0 | 1 | 0 | 5 |
| RsmH | b0082 | 34.9 | 16S rRNA m4C1402 MTase | rRNA (m4C) | 5 | 0 | 0 | 0 | 7 |
| RsmD | b3465 | 21.7 | 16S rRNA m2G966 MTase | rRNA (m2G) | 2 | 0 | 0 | 0 | 2 |
| RsmB | b3289 | 48.3 | 16S rRNA m5C967 MTase | rRNA (m5C) | 1 | 0 | 0 | 0 | 0 |
| MnmC | b2324 | 74.4 | Bifunctional protein includes tRNA (mnm(5)s(2)U34)-MTase | tRNA (mnm5s2U) | 1 | 0 | 0 | 0 | 0 |
| PrmB | b2330 | 35.0 | 50S ribosomal protein L3 Gln150 MTase | protein (Gln) | 13 | 0 | 0 | 0 | 15 |
| CheR | b1884 | 32.8 | Chemotaxis protein MTase | protein (Glu) | 0 | 0 | 0 | 0 | 1 |
| Cfa | b1661 | 44.9 | Cyclopropane-fatty-acyl-phospholipid synthase | small molecule | 15 | 0 | 0 | 0 | 14 |
| Tam | b1519 | 29.0 | Trans-aconitate 2-MTase | small molecule | 2 | 0 | 0 | 0 | 3 |
| CysG | b3368 | 50.0 | Siroheme synthase includes uroporphyrinogen-III C-MTase | small molecule | 1 | 0 | 0 | 0 | 2 |
| SmtA | b0921 | 29.8 | Protein smtA | (?a) | 7 | 1 | 0 | 0 | 8 |
| MtnN | b0159 | 24.4 | 5′-Methylthioadenosine/SAH nucleosidase | small moleculeb | 36 | 0 | 0 | 0 | 39 |
| GlnA | b3870 | 51.9 | Glutamine synthetase | small moleculec | 90 | 0 | 0 | 0 | 97 |
| RplK | b3983 | 14.9 | 50S ribosomal protein L11 | of protein MTase PrmAd | 2 | 0 | 0 | 0 | 2 |
aNot (fully) characterized
bNo methylation but cleavage of the glycosidic bond of SAH
cNo methylation but binding of SAH into the ATP binding site as shown by CCMS experiments with ATP as competitor (data not shown)
dSubstrate of the 50S ribosomal protein L11 MTase PrmA; reproducible specific identification by CCMS (data not shown)
Table 1: MTases and other selected proteins identified by CCMS experiments. The given numbers denote the unweighted peptide spectral count per protein. Samples are duplicates of those analyzed by SDS-PAGE/silver stain in Figure 2. Much more MTases and other SAH binding proteins are identified in the CCMS assay (A) compared to the pulldown (PD) and SAH specificity is shown by the almost complete absence of these proteins in the competition control (C).
Table 2:
| A | C | PD | CPD | A+PD | |
| A | 111 (64) | ||||
| C | 65 (41) | 107 (46) | |||
| PD | 25 (15) | 23 (13) | 61 (17) | ||
| CPD | 23 (13) | 22 (12) | 20 (14) | 47 (14) | |
| A+PD | 87 (61) | 64 (41) | 23 (14) | 22 (12) | 124 (67) |
Table 2: Total number of identified proteins in the CCMS runs and protein overlap between the runs. The number of proteins identified with at least 2 peptides are given in parentheses. The high reproducibility of the method can be inferred from the high protein overlap (of mainly unspecific proteins) between comparable experiments (A vs. C and especially A vs. A+PD but also PD vs. CPD) especially with the proteins robustly identified with at least 2 peptides. See also Figure 3 for Venn diagrams and Supplementary Table S1 for a list of all identified proteins.
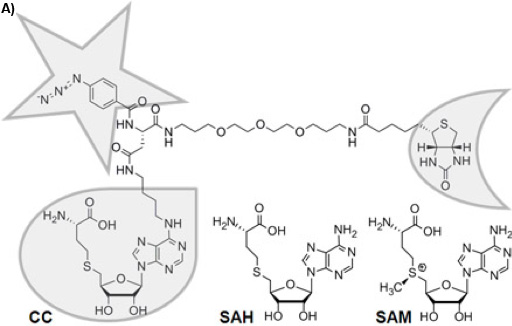
Figure 1A: Chemical structure of the trifunctional Capture Compound (CC). The selectivity function is framed with a droplet, the reactivity function with a star, and the sorting function with a half-moon. The chemically stable S-adenosyl-L-homocysteine (SAH) is the cofactor product of S-adenosyl-L-methionine (SAM) after methyl group transfer by SAM-dependent MTases, for which SAH acts as a product inhibitor.
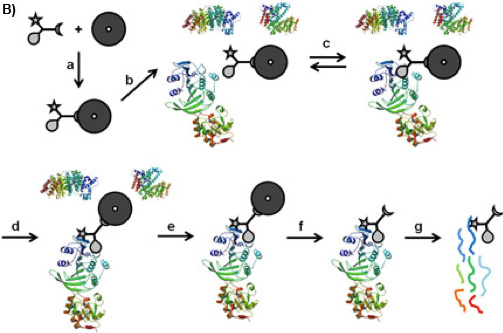
Figure 1B: CCMS “on-bead” workflow. The CC is bound on the magnetic beads by its sorting function (a), the so formed caproBeads are incubated with the complex protein mixture (b), where a reversible binding equilibrium (c) is established between the selectivity function of the CC and the target proteins. Upon UV irradiation (d), the reactivity function forms a covalent crosslink. After washing the magnetic beads bearing the captured proteins (e), cleavage of the crosslinked CC-protein complexes from the magnetic beads (f), and tryptic digest (g), the captured proteins can be identified by MS analysis of the tryptic peptides.
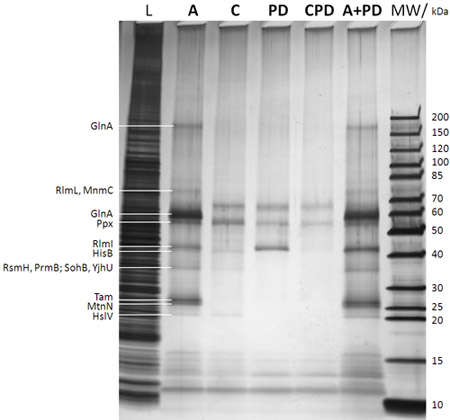
Figure 2: SDS-PAGE/silver stain analysis of captured proteins (after step f in Figure 1B). The lane description is given on top of the gel (MW: molecular weight marker with the corresponding molecular weights of the marker bands given to the very right; L: 0.25% sample drawn from the E. coli DH5a whole cell lysate before adding the caproBeads in step b in Figure 1B; A: assay with addition of an excess of free SAH after UV irradiation step d in Figure 1B; C: control of assay including an excess of free SAH as competitor during steps c and d in Figure 1B (essential to determine any non-specifically captured proteins); PD: pulldown meaning no UV irradiation step d in Figure 1B and no addition of free SAH; CPD: control of pulldown using SAH as competitor; A+PD: combined assay plus pulldown meaning no addition of free SAH during the workflow). Proteins identified by MS from cut-out protein gel bands after in-gel tryptic digest are given to the very leftt. It is evident that photo-crosslinking enhances yield and sensitivity of the experiment, and the specificity can be readily tested for in competition experiments using an excess of free SAH. See Table 1 for MTases and other selected proteins identified by CCMS experiments of duplicate samples of those shown in the present figure.
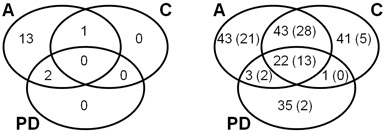
Figure 3: Venn diagrams explaning the overlap of identified proteins in CCMS assay (A), competition control (C), and pulldown (PD). Left: Number of MTases and SAH nucleosidase, only, referring to Table 1. Right: Number of all identified proteins referring to Table 2 and Supplementary Table S1. The number of proteins identified with at least 2 peptides are given in parentheses.
