A. Material Preparation and Sterilization
- Prior to preparing encapsulation materials, prepare a 0.5 wt% solution of the photoinitiator Irgacure 2959 (I2959) in phosphate buffered saline (PBS), by mixing and incubating for several days at 37°C. The solution can then be syringe filtered for sterilization and stored at room temperature for long-term use.
- Based on the desired composition of gels to be synthesized, calculate (using a spreadsheet program such as Microsoft Excel) the weight of polymer and crosslinker needed. Because a given weight of polymer must be dissolved in a defined total volume to achieve a desired concentration in the hydrogels, the crosslinker and polymer solution portions of the total gel volume must also be determined using the spreadsheet. Similar calculations should be made if any pendant moieties (e.g., an adhesive RGD or YGSR containing oligopeptide) are to be incorporated prior to crosslinking.
- Weigh the required amount of polymer and crosslinker and place into Eppendorf tubes. Adjust the calculation spreadsheet accordingly for the actual weight of polymer obtained. (Note: the volumes can be altered to compensate for the actual mass of polymer and crosslinker obtained).
- Prepare gel molds by using a razor blade to cut the tops off of individually wrapped sterile 1 mL disposable syringes. Ensure a flat cut is made for molds to be used for photopatterned gels.
- Place the syringe molds, Eppendorf tubes (with caps open) and any other necessary materials beneath a germicidal light source (typically built into biological safety cabinets) and sterilize for 30 minutes.
B. Cell Preparation
- Following sterilization, add the appropriate sterile buffer volume (per spreadsheet calculations; while the choice of buffer can vary, PBS and a weak base buffer such as triethanolamine are typically used for free radical and michael-type crosslinking, respectively) to the polymer Eppendorf and (optionally) pendant moiety to be included (e.g., adhesive peptide) and vortex to dissolve (if vortexing outside the sterile hood wrap the Eppendorf with parafilm).
- Add the appropriate volume of the pendant moiety solution to the polymer solution and wrap the seal with parafilm. Briefly vortex and incubate at 37°C to react (if possible, atop a rotary stir plate) during cell preparation. This involves the reaction of a thiol on a peptide (via cysteine group) to an acrylate or methacrylate so that the peptide is included as a pendant group in the final hydrogel.
- Trypsinize and count cells and separate into portions containing the appropriate number for each set of gels. For example, for a set of three 50 μL gels at a density of 10 million cells/mL, separate 1.5 million cells into an individual conical tube. It is recommended that no more than 4 gels be prepared in an individual set due to the timing of gelation (note: if synthesizing multiple sets of gels, a single Eppendorf containing an adequate volume of polymer solution for all sets can be prepared).
- Centrifuge the cells.
- While cells are spinning down
- Light initiated free radical crosslinking: add 0.5 wt% I2959 solution to the polymer solution equal to 10% of the total gel set volume (for a final initiator concentration of 0.05 wt%).
- Michael-type (addition) crosslinking: add buffer to the crosslinker to achieve the appropriate concentration solution.
- For sequential crosslinking: these gels incorporate both types of crosslinking and require both of the previous steps.
C. Hydrogel Formation
- Aspirate supernatant from the centrifuged cell portion. Wait for liquid to settle after initial aspiration and re-aspirate to remove as much media as possible without disrupting the cell pellet.
- Resuspend the cell pellet with the polymer solution until cells are evenly distributed. Minimize bubble formation by slowly pipetting the solution up and down (roughly 10 cycles should be sufficient to distribute cells).
- Crosslinking Hydrogels
- Light initiated free radical crosslinking: Aliquot the appropriate volume into syringe tip molds. Expose the syringes to UV light for radical crosslinking (e.g., a 10 minute exposure to 365 nm, 4 mW/cm2 UV light is common for acrylate functionalized polymers). This step should be performed rapidly to minimize cell settling before UV light exposure.
- Michael-type (addition) crosslinking: Using a wide orifice pipette tip, add the appropriate volume of crosslinker solution to the polymer/cell solution, set the pipette to the desired individual gel volume, pipette the solution up and down to homogenize, and aliquot into the syringe tip molds. This step should be performed rapidly to ensure an even distribution of cells. Allow 10-30 minutes (depending upon the individual system) incubation in the culture hood for crosslinking.
- Sequential crosslinking: This involves the Michael-type addition reaction first and then exposure to light for the second crosslinking. Only a fraction of theoretical crosslinks should be reacted during the first crosslinking and then a sterile mask should be applied directly to the gel surface prior to light exposure using sterilized tweezers. A functionalized dye (e.g., methacrylated rhodamine (MeRho) at a final concentration in the gel of 20 nM) may also be added to the initial solution to visualize the patterning, since it will incorporate preferentially in radically crosslinked regions of the gel (Figure 2).
D. Cell Culture and Analysis
- Following crosslinking, plunge gels into wells of a tissue culture plate containing media for incubation and analysis (see following steps).
- Cells can be visualized for morphology using light microscopy (Figure 3; typically, synthesized hydrogels are transparent) and viability using a fluorescent live/dead staining kit (Figures 3 and 4). Staining for cellular actin (e.g., rhodamine or fluorescein labeled phalloidin) and nuclei (e.g., 4’-6-Diamidino-2-phenylindole (DAPI)) can be performed using standard fixation and permeabilization procedures (Figure 4) with the following procedure.
- Rinse constructs 3X for 5 min with PBS.
- Fix gels by incubation in 1 mL 4% formaldehyde for 30 min at room temperature.
- Rinse constructs 3X with blocking solution containing 3% (w/v) BSA and 0.5% (w/v) Tween in PBS.
- Permeabilize membrane with 0.25% (w/v) Triton X in blocking solution for 20 min.
- Rinse constructs 3X with blocking solution.
- Stain for F-actin with 0.66 μg/mL FITC or rhodamine phalloidin in blocking solution for two hours at room temperature.
- Rinse 3x with blocking solution.
- Stain cellular nuclei using 0.4 μL/mL DAPI in PBS for 20 min.
- Rinse 3X with PBS.
- Visualize cells using epifluorescent or confocal microscopy.
- Proliferation of cells within gels is commonly assessed using commercially available assays (e.g., total DNA or biochemical content). PicoGreen is a commonly used and sensitive fluorescent nucleic acid stain for quantifying double-stranded DNA (Singer et al.). Cells may also be counted and normalized to an initial control number using the visualization methods described in Step 2.
- Histological sectioning and staining can also be used to visualize cells and formed matrix. The following protocol for the dehydration and sectioning of 3D hydrogels may be used to obtain cross-sectional slices on glass slides:
- Fixation and dehydration
- Rinse specimen 3X in PBS.
- Fix for 5 – 30 minutes in 4% paraformaldehyde.
- Rinse specimen 3X in PBS.
- Incubate sample in EtOH at 25°C for 1 hour at each of the following EtOH (v/v) concentrations in water (in order): 50%, 70%, 95%, 95%, 100% (At this point, samples can be stored overnight at 25°C).
- Clearing and Infiltration
- Transfer to 100% Citrisolv for 1 hr at 25°C, followed by one hour at 65°C.
- Cut samples in half with a razor blade in a Petri dish such that the cross section of the sample is exposed.
- Transfer to 1:1 mixture of Citrisolv/Micro-cut Paraffin for 1 hr at 65°C.
- Transfer to 100% Micro-cut Paraffin for 1 hr at 65°C.
- Transfer to 100% Micro-cut Paraffin overnight at 65°C.
- Transfer to 100% Micro-cut Paraffin for 1-2 hours at 65°C.
- Embedding
- Position specimen in peel-away mold with 100% paraffin.
- Place a small amount of wax in mold bed, and insert the two halves of the sample with the cross-section facing down.
- Apply top filter piece
- Add additional wax to submerge the top piece and allow to harden for ~10 minutes.
- Transfer inserts to 4°C fridge to allow specimen to harden overnight.
- Specimen may be sectioned the following day, typically at 5-10 μm thickness per slice using commercially available microtomes. Typically the peel-away mold is mounted vertically against the microtome stage, and manual controls are used to set the section thickness, slicer and alignment of the sample with the blade.
- RNA Extraction
Gene expression may also be assessed quantitatively (e.g., via real-time polymerase chain reaction (PCR)). The protocol for RNA extraction from 3D hydrogels, an example of which is given below, is quite different from the much simpler 2D case.- Label a certified RNAse free Eppendorf tube for each 3D hydrogel and add 500 μL Trizol® reagent to each tube.
- Use a pestle (tissue grinder) to manually grind the hydrogels until a clear solution is obtained. Ensure different grinders are used for each condition.
- Vortex each Eppendorf for 20 seconds to homogenize the samples.
- Incubate samples at room temperature for five minutes to permit complete dissociation of nucleoprotein complexes.
- Cool samples on ice for 20 min
- Add 200 μL (per mL Trizol) chloroform to each sample and vortex.
- Incubate samples at room temperature for five min.
- Centrifuge samples at 12,000 x g at 4°C for 15 min.
- Transfer the aqueous phase (clear top layer) to new Eppendorf tubes (avoiding interfacial region and lower red, phenol-chloroform phase). RNA remains in the clear aqueous phase.
- Common techniques for RNA isolation and storage can now be used.
- Following RNA extraction, commercially available kits for reverse transcriptase and real-time PCR are often used with some modifications (Elisseff et al., Strehin et al.).
- Fixation and dehydration
Representative Results:
See Figures 2-4 (as referenced in the above protocols) for representative results of visualization of photopatterned hydrogels and encapsulation of cells within gels.
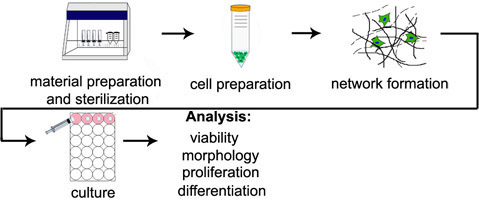
Figure 1. Overview of 3D encapsulation procedure. Diagram steps correspond to section titles in the stepwise protocols.

Figure 2. Photopatterned hydrogel using sequential Michael-type addition then radical polymerization. (A) Schematic of photopatterning of a partially crosslinked hydrogel with incorporation of a photo reactive dye. (B) Circle or (C) stripe transparency film mask patterned hyaluronic acid hydrogels. Methacrylated rhodamine (MeRho) is incorporated only in regions of the gel that were exposed to light. Scale bars = 100 μm.
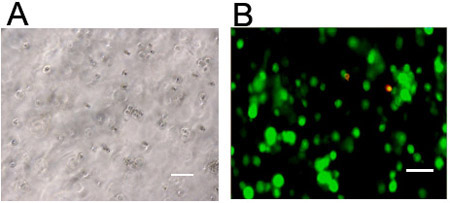
Figure 3. Visualization of cells in 3D hydrogels. Stem cells visualized via (a) light microscopy or (b) live (green, Calcein AM) and dead (red, Ethidium homodimer-1) staining 24 hours following encapsulation at 1 million cells/mL in a hyaluronic acid hydrogel crosslinked through a light initiated free radical mechanism. Scale bars = 100 μm.
Figure 4. Visualization of cells in 3D hydrogels. hMSCs stained for (a) live cells (Calcein AM) or (b) cellular actin (rhodamine phalloidin) and nuclei (DAPI) five days following encapsulation at 1 million cells/mL in a hyaluronic acid hydrogel crosslinked through a Michael-type mechanism (using pendant adhesive peptides and bifunctional proteolytically degradable peptide crosslinkers). Scale bars = 100 μm.
