1. Preparation of 20-kb linear forked DNA used in single-molecule assays (Figure 1)
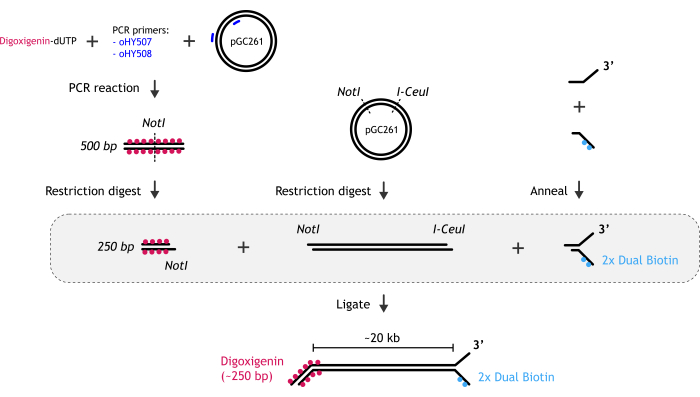
Figure 1: Graphical representation of DNA substrate preparation. (A) The biotinylated DNA fork end is created by annealing two partially complementary oligonucleotides: biotinylated and non-biotinylated one. (B) The main dsDNA fragment (~20 kb) is generated by restriction digest of pGC261 plasmid with two enzymes to create a linear DNA with different overhangs at each end. (C) The digoxigenin duplex DNA end is obtained by a PCR reaction performed in the presence of digoxigenin-dUTP, followed by restriction digest. Please click here to view a larger version of this figure.
- Generation of biotinylated DNA fork end.
- Anneal the fork DNA.
- Mix two oligonucleotides (oHY502 and oHYbio85) in 1x STE buffer (100 mM NaCl, 10 mM Tris, pH 8.0; 1 mM ethylenediaminetetraacetic acid [EDTA]). Final oligonucleotide concentration is 10 µM, and the final reaction volume is 100 µL.
- Incubate the reaction at 80 ˚C for 5 min on a heat block. Switch off the heat block and allow the reaction to cool gradually, leaving it in the heat block until the temperature drops below 30 °C.
- Validate the annealing efficiency on a Tris-Borate-EDTA (TBE)- Polyacrylamide gel electrophoresis (PAGE) gel.
- Prepare 8% acrylamide – TBE gel (for 4 gels: 32.8 mL of H2O; 4.8 mL of 10x TBE buffer (1 M Tris, 1 M boric acid, 20 mM EDTA), 9.6 mL of 40% Acrylamide/Bis Solution, 37.5:1; 0.8 mL of 10% Ammonium persulfate [APS], 40 µL of Tetramethylethylenediamine [TEMED]).
CAUTION: TBE, acrylamide, APS, TEMED are toxic/harmful. - Run a small volume (1 µL) of annealed oligonucleotides alongside the individual oligonucleotides diluted with 6x loading dye on the 8% acrylamide – TBE gel at 80 V, for 1 h at RT.
- Soak the gel in 1x TBE supplemented with a suitable nucleic acid stain for 30 min on a see-saw rocking shaker, then scan the gel using a gel imaging system.
- Prepare 8% acrylamide – TBE gel (for 4 gels: 32.8 mL of H2O; 4.8 mL of 10x TBE buffer (1 M Tris, 1 M boric acid, 20 mM EDTA), 9.6 mL of 40% Acrylamide/Bis Solution, 37.5:1; 0.8 mL of 10% Ammonium persulfate [APS], 40 µL of Tetramethylethylenediamine [TEMED]).
- Resolve annealed DNA on TBE-PAGE gel for gel excision.
- If the oligonucleotides are annealed properly, resolve the rest of the sample in the presence of BSA (~100 µL of annealed sample, 25 µL of 6x loading dye, 2 µL of 33 mg/mL BSA). To load the larger volume on the gel, combine several wells with a sterile scalpel.
- Leave the gel in 1x TBE supplemented with suitable nucleic acid stain for 30 min on a see-saw rocking shaker.
- Transfer the gel into a glass tray and visualize the DNA in a dark room under blue light. Use a suitable protective screen or protective glasses when using blue light.
- Cut out the desired band with a sterile scalpel. Trim off excess gel.
- Purify the annealed fork by electroelution.
- Cut a piece of dialysis tubing long enough to insert the gel piece, wet it in 1x TBE, and clip one end shut.
- Dilute BSA to 0.3 mg/mL in 500 µL of 1x TBE and pipette the whole volume into the tubing.
- Use the metal spatula to slide the gel piece inside. Move the gel piece to one side of the tubing to give buffer space for the DNA to migrate into during the electroelution.
- Squeeze out the excess buffer and clip the second end of the tubing.
NOTE: The remaining volume will determine the final volume and concentration. - Place the dialysis tubing into the agarose electrophoresis tank in 1x TBE buffer, ensuring the tubing is fully submerged.
NOTE: Ensure that the gel piece is placed on the side closest to the negative electrode so DNA can migrate out of the gel into the dialysis bag. - Run the electroelution at 80 V for 1-2 h.
- Dialyze the electroeluted DNA into 10 mM Tris buffer (pH 8).
- After electroelution, take the dialysis bag out of the tank and dry one end with tissue. Avoid touching the middle part of the tubing with tissue, where the gel and the sample are located.
- Remove the clip from the dried end and gently pipet the sample up and down inside the tubing to mix the DNA into the buffer.
- Dip a clean spatula into 1x TBE buffer and scoop out the gel piece from the tubing.
NOTE: While removing the gel piece, remove as little buffer as possible. - Clip the open end of the dialysis bag shut again, removing any air from the inside.
- Place the dialysis bag into a 2 L beaker filled with 1.5 L of 10 mM Tris buffer (pH 8), 20 mM NaCl, and 2 mM MgCl2. Wrap the clips with parafilm to stick the tubing to the edge of the beaker while fully submerging the tubing in the buffer.
NOTE: Salt stabilizes short oligo duplexes. - Use a magnetic bar to gently stir the buffer and dialyze the sample at room temperature (RT) for at least 3-4 h or at 4 ˚C overnight.
- After dialysis, dry one end of the dialysis bag with a tissue (not to dilute the DNA) and unclip this end.
- Gently pipette the sample up and down inside the tubing and transfer it into a clean 1.5 mL tube.
- Measure the concentration of the DNA using a microvolume spectrophotometer.
- Anneal the fork DNA.
- Generation of 20-kb fragment
- Perform restriction digest of pGC261 plasmid18 using NotI/I-CeuI enzymes.
- Gently mix 8 µL of NotI-HF (20,000 U/mL), 8 µL of I-CeuI (5,000 U/mL), and the pGC261 plasmid (final plasmid concentration ~40 ng/µL in a 200 µL reaction) in a buffer recommended by restriction enzymes producers.
- Incubate the reaction at 37 ˚C overnight.
- Resolve digested DNA on agarose gel for gel excision.
- OPTIONAL: Run a small amount of the reaction on a 0.6% agarose gel first to test digestion efficiency before loading the whole reaction.
- Prepare 0.6% agarose gel following steps 1.2.2.3-1.2.2.4.
- Mix 0.48 g of agarose powder with 80 mL of 1x TBE buffer. Microwave the solution until it is boiling. Swirl to ensure that the agarose is fully melted. Allow to cool for a few minutes, then pour into the designated tank.
- To combine wells to accommodate the larger volume of the sample, stick a piece of tape across multiple wells of the comb beforehand.
- Once the gel has solidified, gently remove the comb and top up the tank with 1x TBE buffer. Gently mix the sample with DNA loading dye and run the gel at 120 V for 1 h.
- Leave the gel in 1x TBE supplemented with a suitable nucleic acid stain on a see-saw rocking shaker until stained.
- Transfer the stained gel to a glass tray and visualize the DNA in a dark room under blue light.
- Cut out the desired band with a sterile scalpel. Trim off excess gel.
- Optional: Purify the desired fragment by electroelution as described in steps 1.1.4 and 1.1.5. Omit the salt from the dialysis buffer here, as this is only needed to stabilize the short oligo duplex.
- Perform restriction digest of pGC261 plasmid18 using NotI/I-CeuI enzymes.
- Generation of digoxigenin duplex DNA end
- Perform PCR reaction with digoxigenin-dUTP (dig-dUTP) using pGC261 DNA as the template.
- Mix: 400 µL of water, 8 µL of pGC261 (0.8 ng/µL), 3.5 µL of primer oHY507 (100 µM), 3.5 µL of primer oHY508 (100 µM), 8 µL of dig-dUTP (1 mM), 400 µL of 2x PCR mix (prepared from DNA polymerase (20 µL), 10 mM dNTPs (40 µL), 5x high-fidelity (HF) buffer (400 µL) and water (540 µL).
- Run the following reaction in a thermal cycler:
98 ˚C – 1 min
30x: 98 ˚C – 20 s; 65 ˚C – 20 s; 72 ˚C – 30 s;
72 ˚C – 10 min
4 ˚C – Hold
- Digest and purify digoxigenin-labeled PCR product
- Purify PCR product using a commercial DNA purification kit.
- Mix 10 µL NotI-HF (20,000 U/mL) with the PCR product in a buffer recommended by the enzyme supplier (final DNA concentration of ~50 ng/µL in a ~200 µL reaction). Incubate the reaction at 37 ˚C overnight.
- Purify the digested DNA using a commercial DNA purification kit.
- Perform PCR reaction with digoxigenin-dUTP (dig-dUTP) using pGC261 DNA as the template.
- Assemble components to make the DNA substrate.
- Gently mix the biotinylated fork end, 20-kb DNA fragment, and digoxigenin-labeled Not-I treated PCR fragment with 5 µL of T4 DNA ligase (400,000 U/mL) in a buffer recommended by the enzyme supplier in a 200 µL reaction. However, based on the 20-kb DNA fragment added (1-5 µg), add approximately a 100-times molar excess of both the biotinylated fork end and PCR fragment.
- Aliquot the reaction into PCR tubes (50 µL each) and incubate at 16 ˚C in a thermal cycler overnight.
- Resolve the ligated sample on a 0.6% agarose gel and purify ligated DNA by electroelution as described in steps 1.2.2 and 1.2.3.
- Store flash frozen DNA at -80 ˚C.
2. Purification of Drosophila melanogaster CMG (Figure 2)
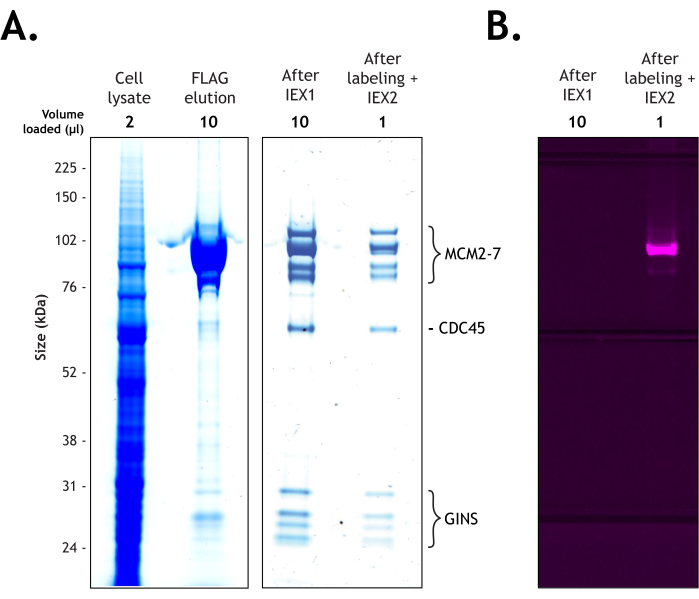
Figure 2: Purification of Drosophila melanogaster CMG from 4 L of Hi Five cells. The proteins were resolved on 4%-12% Bis-Tris polyacrylamide gel under 200 V in the presence of MOPS buffer. The sample is shown at each stage of the purification (cell lysate – 2 µL, FLAG elution – 10 µL, after the first ion exchange column – 10 µL, and after labeling and the second ion exchange column – 1 µL. (A) Coomassie staining confirms the presence of all 11 subunits of the CMG complex before (10 µL), and after (1 µL) fluorescent labeling. (B) The labeling efficiency of the MCM3 subunit was validated by scanning for Cy5 with a fluorescent image analyzer using a long pass red (LPR) filter. Please click here to view a larger version of this figure.
NOTE: To prepare fluorescently labeled Drosophila melanogaster CMG, a TEV cleavage site (ENLYFQG) followed by four Gly residues was introduced downstream of the N-terminal FLAG Tag on the MCM3 subunit (in pFastBac1 vector)10. To express the complex, the baculovirus expression system was used. For initial transfection, Sf21 cells were used separately for each CMG subunit (P1 virus stage). To further amplify the viruses, Sf9 cells were used (P2 virus stage). Subsequently, Sf9 cell cultures (100 mL for each CMG subunit; 0.5 x 106 cells/mL) were infected with 0.5 mL P2 virus supplemented with 10% fetal calf serum (P3 virus stage). To express the entire CMG complex in 4 L of Hi Five cells (1 x 106 cells/mL), 200 mL of P3 viruses were used for each of the subunits. After harvesting the Hi Five cells expressing the CMG complex, the cell pellet can be flash frozen in liquid nitrogen and stored at -80°C. Perform the whole purification on ice or at 4 °C. The buffers can be prepared in advance, providing that the reducing agents (DTT or 2-Mercaptoethanol) and protease inhibitors (CAUTION) are added just before the use. Ensure that all buffers are precooled in advance, filtered, and degassed.
- Prepare the following buffers.
- Prepare the resuspension buffer A by mixing 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5, 0.02% Tween-20, 10% glycerol, 15 mM KCl, 2 mM MgCl2, 2 mM 2-mercaptoethanol, 1 mM EDTA, and 1 mM ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA).
CAUTION: 2-mercaptoethanol, DTT, and EDTA are toxic/harmful - Prepare Tris-buffered saline (TBS; 0.1 M Glycine HCl, pH 3.5) buffer for preparing ANTI-FLAG M2 affinity gel.
- Prepare Buffer A-100 by mixing 25 mM HEPES pH 7.5, 0.02% Tween-20, 10% glycerol, 100 mM KCl, 1 mM DTT, 1 mM EDTA, and 1 mM EGTA.
- Prepare dialysis buffer by adding 25 mM HEPES pH 7.5, 50 mM sodium acetate, 10 mM magnesium acetate, 10% glycerol, and 1 mM DTT.
- Prepare TBS buffer by mixing 50 mM Tris-HCl pH 7.5 and 150 mM NaCl.
- Prepare the resuspension buffer A by mixing 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5, 0.02% Tween-20, 10% glycerol, 15 mM KCl, 2 mM MgCl2, 2 mM 2-mercaptoethanol, 1 mM EDTA, and 1 mM ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA).
- Resuspend pellet from 4 L culture of Hi Five cells in 200 mL of cold resuspension buffer A supplemented with protease inhibitor cocktail tablets. Mix the tubes by inversion until the entire pellet is fully resuspended.
- Transfer the resuspended cells into precooled 40 mL Dounce homogenizer. To recover as many cells as possible, wash the tubes used for the cell pellet freezing with the same type of buffer and add to the Dounce homogenizer. Ensure that the total volume in the homogenizer does not exceed the recommended limit. Lyse the cells on ice by 60-70 strokes.
NOTE: Ensure that the plunger reaches the bottom of the tube, but do not put too much pressure as the homogenizer may break. Be careful not to take out the plunger above the liquid level, as this will introduce bubbles. - Repeat the previous step until the entire sample is homogenized and combine it in the precooled beaker. Assess the final volume of the sample. Drop by drop, add filtered KCl to the final concentration of 100 mM, gently mixing.
- Precool the centrifuge to 4 ˚C in advance. Pour the sample into centrifugation tubes, ensuring the volume recommended by the producer is reached. Balance the tubes on a scale.
NOTE: Tubes with too small a volume may break during centrifugation. - Centrifuge the lysed cells at 23,500 x g for 15-30 min at 4 °C. In the meantime, prepare ANTI-FLAG M2 affinity gel in line with the next step.
- Gently mix the bottle containing ANTI-FLAG M2 affinity gel (50% suspension). Cut the end of a P-100 pipette tip (to widen tip) and use it to immediately transfer 4 mL of suspension (2 mL of beads) into a 20 mL poly-prep chromatography column. To recover the beads stuck in the tip, wash the tip with TBS and add it to the column. Allow all the beads to settle inside the column, but be careful not to dry them.
- Wash the resin with 3x beads volume (6 mL total) of 0.1 M Glycine HCl pH 3.5. Subsequently, wash the beads with 3-5 beads volume of TBS (6-10 mL). Next, wash the beads 3 times (6 mL) with Buffer A-100.
NOTE: Do not leave the beads in the 0.1 M Glycine HCl pH 3.5 solution for longer than 20 min. To increase the flow pressure and washing speed, cover the column with a lid. Do not allow the resin to dry at any point. - After preparing the resin, leave 2 mL of Buffer A-100 above the resin and close the outlet of the column.
- After the centrifugation, gently pour the supernatant into precooled 50-mL tubes. Be careful not to disrupt the pellet. Take a small aliquot (~10 µL) to validate by SDS-PAGE electrophoresis later.
- Add an equal amount of ANTI-FLAG M2 beads (prepared in line with the previous step) into each of the 50-mL tubes. Try to recover all the beads from the column by resuspending them with a couple of additional milliliters of Buffer A-100 and transferring this resuspension to the tubes.
- Incubate the sample (supernatant) with ANTI-FLAG M2 resin, rotating for 2.5 h at 4 °C.
- After the incubation, spin the sample at 1,000 x g for 5 min at 4 °C. Using a pipette, remove the majority of the supernatant. Using a cut tip, resuspend the beads in few milliliters of the remaining supernatant and transfer them into two 15-mL tubes. To recover any beads stuck to the walls of the 50-mL tubes, add a few milliliters of Buffer A-100 and transfer this to the 15-mL tubes as well.
- Spin the 15-mL tubes at 1,000 x g for 5 min at 4 °C. Remove the supernatant.
- Wash the beads by adding ~14 mL of Buffer A-100 to each tube, followed by short, gentle rotation at 4 °C and subsequent spinning at 1000 x g for 5 min at 4 °C. Repeat the washes twice. Take small aliquots of each wash (~10 µL) to validate by SDS-PAGE electrophoresis later.
- This step (2.16) is an alternative procedure to the previous steps (2.13-2.15).
- After the incubation with ANTI-FLAG M2 resin, pour the sample into two 20 mL poly-prep chromatography columns.
- If choosing this method, let the beads settle at the bottom of the column and the unbound sample go through the column by the gravity flow. Subsequently, add Buffer A-100 directly to the column and let it wash the sample via gravity flow.
- Transfer the beads (resuspended in Buffer A-100) to two 10-mL poly-prep chromatography columns.
- Allow Buffer A-100 to pass through the resin, close the outlet when the buffer level reaches the top of the resin, and elute the protein from the beads by adding Buffer A-100 supplemented with 200 µg/mL (DYKDDDDK) peptide (FLAG elution buffer).
- For the first elution, add 3 mL of the FLAG elution buffer and gently rotate the closed column for 15 min at RT. Open the outlet to collect the first elution fraction.
- Close the outlet and add 2 mL of the FLAG elution buffer to each column. Rotate for another 10 min and collect the second elution fraction.
- Combine all the eluted fractions together (~10 mL) and keep at 4 °C. Take a small aliquot of the eluted fraction (~10 µL) to validate by SDS-PAGE electrophoresis later.
- Filter the eluted sample using 0.22 μm syringe filters.
- In advance, prepare the CaptoHiRes Q (5/50) column (high-resolution ion exchange chromatography column) connected to a protein purification system, in line with the manufacturer's protocol. Subsequently, equilibrate the column with Buffer A-100 and load the filtered sample onto the equilibrated column.
- Wash the column with 20 column volumes (CV) of Buffer A-100 (~20 mL total as the volume of the column is ~1 mL).
- To elute the protein, prepare two buffers in advance: Buffer A-100 and Buffer B with the same composition as Buffer A-100, but with 550 mM KCl instead of 100 mM KCl. Set up the gradient elution for 20 CV (~20 mL) with increasing salt concentration from 100 mM to 550 mM KCl. Collect 0.3-0.5 mL elutions in a fraction collector.
NOTE: The CMG should elute at around 70%-75% concentration of Buffer B. Take small aliquots of the wash and eluted fractions for SDS-PAGE electrophoresis. - Perform SDS-PAGE electrophoresis to confirm that CMG is present in the chosen fractions. In the meantime, clean the column and purification system per the manufacturer's protocol.
- Incubate the chosen fractions with TEV protease overnight by mixing 50 µL of TEV protease (1 mg/mL) per 1 mL of sample.
- Prewet a dialysis tube and ensure that the membrane is not damaged. Add the CMG/TEV mix into the dialysis tubing and place into 2 L beaker with 1.5 L of precooled Buffer A-100 and the magnetic bar inside. Dialyze with gentle stirring overnight at 4°C.
- Mix the sample with 50 µM of LD655-labeled peptide and 10 µg/mL Sortase enzyme in the presence of 5 mM CaCl2. Incubate the reaction at 4 °C, rotating for 30 min, ensuring the tube is covered from light. Take a small aliquot before and after the labeling for SDS-PAGE electrophoresis.
- Filter the labeled sample using 0.22 μm centrifuge filters before loading onto the purification system, as the peptide may precipitate.
- Prepare the high-resolution ion exchange chromatography column in advance, as described before (step 2.20). This time, cover the column and the fraction collector system with aluminum foil to protect the sample from light.
- To remove the free peptide, load the filtered sample on the high-resolution ion exchange chromatography column. Perform the purification the same way as described before (steps 2.21-2.22). Ensure that the labeled CMG elutes at a concentration of Buffer-B similar to that previously.
- Perform SDS-PAGE electrophoresis to validate the quality of chosen fractions. To image the fluorescence, do not boil the samples before loading, and ensure the electrophoresis tank is protected from light. Visualize protein fluorescence using a gel imaging system first, then stain the gel with Coomassie dye and image it again to visualize all proteins.
- Dialyze chosen fractions overnight at 4 °C, against 1.5 L of the dialysis buffer. If necessary, concentrate the sample.
- Flash freeze the protein in liquid nitrogen and store it at -80 °C until further use.
3. Preparation of the flow cell (Figure 3)
- Prepare biotin-PEG coverslips following a previously described protocol11, omitting the oven baking step. Biotin-PEG coverslips are stable for at least 1 month under vacuum at RT. For preparation of the flow cell, cut the biotin-PEG coverslip (24 mm x 60 mm) in half (approximately 24 mm x 30 mm).
NOTE: Take care not to touch the central area of the coverslip where the flow channels will be. - Prepare small glass pieces by etching and snapping a glass slide into approximately 2.4 cm x 1 cm pieces.
- Drill two holes 1.4 mm apart using a 0.8 mm diamond-coated drill bit, one slightly wider than the other (0.043" = inlet, 0.048" = outlet). Test that the holes are the correct size by trying to insert a piece of the inlet/outlet tubing, using the same drill bit to widen the hole until the tubing fits. Ensure that the tubing fits tightly enough to not fall out easily.
NOTE: Narrower inlet tubing is used to minimize the dead volume. - Cut double-sided tape the same shape as the glass pieces.
- Align the slide on the tape and stab a needle through each hole to mark their positions on the tape. Using a razor blade, cut a channel that encompasses both holes.
NOTE: Do not cut the channel too long, as excess space on the far side of each hole may remain dry when liquid is flowed in and can lead to issues with air bubbles during flow. When cutting, try to cut the long edges in a single clean cut, as frayed edges from multiple cuts can affect the quality of the flow channel. To a similar end, do not cut into the edges of the flow channel, as cuts into the usable piece can affect the flow of liquid or even leak in the assembled flow cell. - Clean the glass piece with acetone and tissue until dry, then place on a clean surface. Peel one side of the adhesive tape and stick it onto the glass piece such that both holes are fully inside the channel.
- Seal the tape to the glass piece by using a p1000 pipette tip to eliminate bubbles by pressing the surface with firm but moderate pressure. Go over the entire surface to seal.
- Peel the second edge of the tape off both glass pieces and place them on a clean surface with the sticky side up. Place the slides close to each other but not touching in the position they will be attached to the coverslip (leave enough space between them for the epoxy to create a full seal around each glass piece).
- Pick up the half biotin-PEG coverslip using plastic tweezers, holding only by the edge (do not touch the area that will form the flow channel), and lower the PEG-functionalized side onto the adhesive. Secure by pressing down with a finger to fix it in place, then complete the seal by rubbing the surface moderately firmly with a pipette tip to remove air bubbles (not on the area of the flow channel as this may break), then flip over.
NOTE: Handle the coverslip with care. - For each flow cell, cut ~10 cm of each type of polyethylene tubing (PE20 and PE60). Use the narrower tube as an inlet to the flow chamber to reduce dead volume.
- Insert tubing into the holes in the slide by hand. If the diameter of the holes is correct, ensure that the tubing stands up in the hole on its own once inserted. Cut the tubing tip at a slight angle <45˚ to make inserting the tubing slightly easier and prevent the tubing from forming a seal against the coverslip, which it may do if the end is flat (this may obstruct flow).
NOTE: The accurate fitting of the tubing into the glass piece makes flow cell construction more reliable and helps avoid the introduction of bubbles to the flow cell. - Mix the epoxy components well, then use a p200 tip to dab epoxy to seal the tubing and create a seal around each glass piece. Add enough epoxy so that it goes up to the edge of the coverslip round, as this will also reinforce the delicate coverslip. Ensure, however, that no epoxy gets onto the underside of the coverslip, as it may prevent it from lying flat on the stage.
NOTE: If necessary, small amounts of excess epoxy can be scraped off the underside using a razor blade. - Leave for at least 30-60 min for the epoxy to fully cure.
NOTE: If the flow cell is not to be used right away, store it under vacuum at RT after fully curing. - After using the flow cell, recover the glass pieces and reuse them indefinitely. Pull the tubing out and place the flow cell into a slide jar containing acetone for at least 24 h to soften the epoxy and double-sided adhesive, allowing the glass pieces to be easily removed. Before reusing to make another flow cell, clean the glass pieces by scrubbing them with a sponge and soapy water, dry them, and scrub them with acetone and tissue paper.
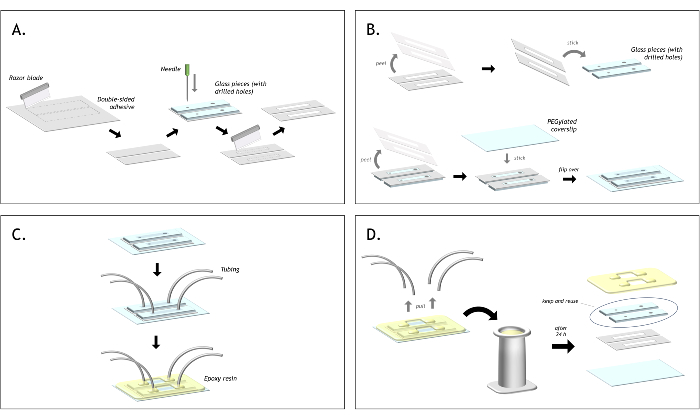
Figure 3: Graphical representation of the flow cell preparation. (A) Cut double-sided tape to match the size of the glass piece. Align the slide on top of the tape and mark the position of each hole with a needle. Using a razor blade, cut around each hold to create a channel. (B) Peel one side of the tape and stick the tape onto the glass piece. Ensure that both holes are inside the channel. Peel the second end of the tape and stick the biotin-PEG coverslip on top. (C) Insert the polyethylene tubing into each hole and seal the tubing in place with epoxy, sealing each glass piece to the coverslip too. (D) After using both channels, pull out the tubing and place the flow cell into a staining jar filled with acetone. After approximately 24 h, the epoxy and tape will have softened, and the layers of the flow cell can be peeled apart. The glass pieces can be recovered and stored in acetone to be reused indefinitely for making the next flow cell. Please click here to view a larger version of this figure.
4. Single molecule TIRF assay to visualize CMG-mediated DNA unwinding
- Prepare the following buffers.
- Prepare Blocking buffer (20 mM Tris, pH 8.0; 50 mM NaCl; 2 mM EDTA; 0.2 mg/mL BSA). Filter the buffer using a 0.22 µm syringe filter and store it at 4 ˚C.
- Prepare 10x Reaction buffer 1 (250 mM HEPES pH 7.5; 50 mM NaCl; 100 mM Magnesium Acetate). Filter the buffer using a 0.22 µm syringe filter and store it at 4 ˚C.
- Prepare 10x Reaction buffer 2 (250 mM Tris, pH 7.5; 100 mM Magnesium Acetate; 1.25 M Potassium glutamate; 1 mM EDTA; 0.025% Tween-20). Filter the buffer using a 0.22 µm syringe filter and store it at 4 ˚C.
- Degas approximately 2.5 mL of blocking buffer and 5 mL of ultrapure water by placing the open tubes in a desiccator and leaving them under vacuum for 15 min.
NOTE: Do not fill tubes to the brim, as bubbles during degassing can cause liquid to splash. - Place the flow cell on a microscope stage and secure it in place using an adhesive putty at each end. It is important to secure well to prevent the flow cell from moving later during the acquisition.
- Connect the flow cell outlet tubing to the syringe pump, which has tubing attached to the syringe with a needle at the other end. Insert this needle into the outlet tubing to connect the flow cell.
- Switch on the objective heater to 30 ˚C.
- Secure 1-2 mL of degassed water in a tube to a separate piece of adhesive putty near to the flow cell such that the inlet tubing can be inserted and reach the bottom of the tube.
- Flow water through the channel, then use faster flow to remove any trapped bubbles near the inlet tubing, adding more water if necessary. Otherwise, large bubbles may become dislodged later in the experiments and pass through the channel. Leave for a few minutes for the flow to stop fully, as the fast flow can cause residual pulling of liquid for a while after it is stopped, as the pressure inside the syringe stabilizes.
NOTE: Any DNA that contacts a bubble becomes unusable, even once rehydrated. - Add 100 µL of degassed blocking buffer to a 20 µL aliquot of 1 mg/mL streptavidin. Attach the open tube to the adhesive putty and transfer the inlet tubing from the water to the streptavidin. Flow at a rate of 40 µL/min for 2 min (80 µL total) and incubate for 5 min.
- Wash out excess streptavidin with the blocking buffer (50 µL/min for 100 µL).
- Flow in biotinylated DNA, diluted in blocking buffer with 25 nM SYTOX orange. Image using live view with the 532 nm laser to watch the DNA tethering to the surface in real-time.
- When the approximate density of DNA on the surface is achieved, flow in blocking buffer, also with 25 nM SYTOX (50 µL/min for 100 µL), to wash out the free DNA.
- Flow in biotinylated anti-digoxigenin antibody (~10 µg/mL) diluted in blocking buffer containing 25 nM SYTOX orange (100 µL/min for 300 µL).
NOTE: The SYTOX orange intercalates between the base pairs of DNA, extending the contour length. This means the end-to-end length of the DNA increases in SYTOX orange, allowing the second tether to attach to the surface further from the first, giving better stretched DNA when the SYTOX orange is washed out later. Using a different moiety on each DNA end allows more uniform tethering of DNA to the coverslip surface (Figure 4). When both ends are biotinylated, they tether in the same step, resulting in significant variation in their attachment positions. Using digoxigenin at one end gives more control over DNA coverage before tethering the second end. This also allows DNA coverage to be increased by incubation. - Wash out the biotinylated anti-digoxigenin antibody and SYTOX orange with blocking buffer (50 µL/min for 100 µL).
- Make up 120 µL of 'ATP-g-s mix' (1x Reaction buffer 1, 0.75 mg/mL BSA, 1.25 mg/mL casein, 8 mM DTT, 0.33 mM ATP-g-s), add 30 µL to a new tube, then flow 50 µL from the original tube into the flow cell (50 µL/min for 50 µL). This helps to minimize the effect of buffer mixing through backflow or diffusion from the outlet tubing.
- Add purified CMG to ~100 nM final in 30 µL of ATP-g-s mix, then flow in at 20 µL/min for 20 µL. Incubate for 15 min.
- Make up 120 µL of 'ATP/RPA mix' (1x Reaction buffer 1, 0.75 mg/mL BSA, 8 mM DTT, 3.3 mM ATP, 20 nM EGFP-hRPA), and flow into flow cell at 40 µL/min for 80 µL.
- Immediately start acquiring images. Acquire 6 x 6 fields of view for each frame every 30 s (or an appropriate frame rate for the experiment). Lower frame rates can reduce laser exposure and fluorophore photobleaching. Visualize EGFP-hRPA with a 488 nm laser at 1% power. If CMG is labeled, e.g., LD655, visualize CMG with 640 nm laser (max power = 30 mW) at 10% power. Visualize SYTOX orange-stained DNA with a 532 nm laser at 0.5% power. Use each laser with 50-100 ms exposure.
NOTE: Each time the inlet tubing of the flow cell is transferred from one tube of liquid to another, dab the tip of the tubing on the bottom of the tube several times before removing it from the liquid. This helps to prevent bubbles from getting in during transfer. Minimize the time the tip of the inlet tubing is held out of liquid, and do not point the tip upwards or raise the tip significantly, as this causes the liquid to drain towards the channel and pull air into the tip. While transferring the inlet tubing, try not to put tension on the tubing as this can pull it out of the epoxy, securing it to the flow cell. For imaging DNA unwinding, in the presence of SYTOX orange to visualize double-stranded DNA, replace 10x Reaction buffer 1 with 10x Reaction buffer 2 for the ATP/RPA mix. SYTOX orange binds to DNA better in these conditions. Fully empty the syringe pump at the end of each experiment. Wash occasionally by filling with water and emptying several times or dismantling and washing with soapy water.
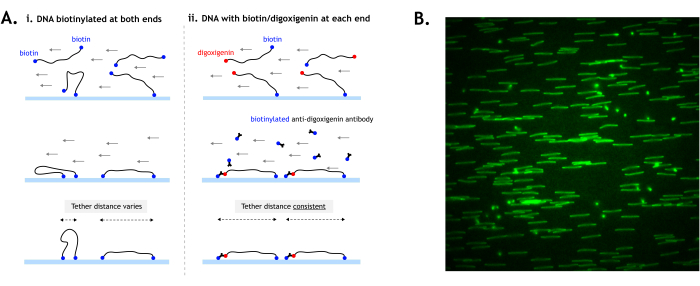
Figure 4: DNA tethering to the surface. (A) When tethering DNA substrates with biotin at both ends, the distance between the two tethers can vary depending on how the ends contact the surface (i). By using digoxigenin at one end, the tethering of each end can be temporally separated for more consistent tether distances and more uniformly stretched DNA (ii). (B) Example field of view showing DNA tethered by both ends (digoxigenin-labeled) and stained with fluorescent intercalating nucleic acid stain. DNA, which is tethered by both ends, appears as a line, while DNA tethered by only one end appears as spots. Ideally, the DNA should be tethered as densely as possible without overlapping other DNA. The image is 512 x 512 pixels (pixel size = 154.6 nm). Please click here to view a larger version of this figure.
When CMG unwinds DNA, a characteristic RPA tract will grow over time (Figure 5). The 5' end of the unwound DNA is tethered to the surface; hence, it is seen as a linear stretch of RPA signal between the tether and the fork. The 3' end is not tethered and, therefore, moves with the fork and is observed as a compact EGFP-RPA signal. The position of the compacted unwound translocation strand corresponds approximately to the position of the replication fork, which moves together with LD655-CMG visualized via a 640 nm laser.
It is important to minimize damage to the DNA substrate, as damage such as single-stranded DNA nicks reduces the number of observable unwinding events, limiting the amount of data that can be collected (Figure 6).

Figure 5: Single-molecule DNA unwinding assay. The DNA substrate is tethered to a coverslip surface. Purified CMG labeled with LD655 is incubated with the DNA for 15 min in ATP-g-s. ATP and purified EGFP-labelled RPA are added, initiating extensive DNA unwinding by CMG. A cartoon schematic (left) and kymograph of representative data (right) are shown. Please click here to view a larger version of this figure.

Figure 6: DNA damage reduces assay throughput. (A) CMG cannot unwind DNA past a break in the DNA backbone (DNA nick). A nick on the leading strand template causes CMG to slide off the DNA, and both CMG and leading strand template are lost. A nick on the lagging strand template causes the lagging strand template to separate from the rest of the DNA, and each DNA piece retracts to their respective tether. This is illustrated by (i) cartoon schematics and (ii) kymographs of these events (ii). Representative data with (B) a minimally damaged DNA substrate versus (C) a more damaged DNA substrate at (i) 5 min, (ii) 15 min, and (iii) 60 min in a single field of view. The more damaged DNA substrate does not generate long tracts of unwinding, as the CMGs encounter nicks earlier on, despite similar levels of unwinding activity (similar density of growing RPA spots at 5 min, indicating similar CMG loading/unwinding efficiency). The field of view is 512 x 512 pixels (pixel size = 154.6 nm). 1% laser power (488 nm) imaging EGFP-RPA. Scale bar showing 10 µm. Please click here to view a larger version of this figure.
