GpsB phenotypes
Previously we have shown that Sa-GpsB is an essential protein as depletion of GpsB using an antisense RNA results in cell lysis9. Here we describe how the emergence of various cell division phenotypes and changes in protein localization could be captured using the timelapse microscopy protocol described in this article. For this purpose, S. aureus strains RB143 [SH1000 harboring pEPSA5 (empty vector)] and GGS8 [SH1000 harboring pGG59 (Pxyl-gpsBantisense bla cat)] reported previously9, were grown as follows. Strains RB143 and GGS8 were inoculated in 2 mL of tryptic soy broth (TSB) supplemented with 5 µg/mL chloramphenicol (chlor) in a 15 mL test tube and were incubated overnight at 22 °C while shaking. The overnight cultures were diluted 1:20 in 10 mL of fresh TSB + chlor in a 125 mL flask and grown at 37 °C with shaking until mid-logarithmic phase (OD600 = 0.5). The inducer, 1% xylose, was added to the culture medium to trigger the expression of antisense RNA of gpsB and the culture was grown for another 3 h. Cells were then stained with fluorescent dye FM4-64 (membrane stain), where required, by the addition of 0.5 µL of a 10 µg/mL stock of FM4-64 directly onto the 5 µL aliquot of culture on the microscope dish as described in the protocol section. As shown in Figure 1 and Video 1, addition of xylose to induce GGS8 strain resulted in a “sick” cell phenotype, as described previously9, while empty vector control (RB143) appeared similar to our control—cells grown in the absence of inducer.
Our group also reported that overproduction of S. aureus GpsB (Sa-GpsB) disrupts cell division in B. subtilis9. We use this overexpression phenotype as an example to demonstrate the protocol described here. To this end, a B. subtilis strain GG9 (amyE::Phyperspank-gpsBSa spc; ftsAZ::ftsAZ-gfpΩerm) was used9. Subcellular localization of fluorescently-labeled FtsZ, a key cell division protein which marks the cell division sites10,11, was used to monitor the status of cell division. The sample for microscopy was prepared as follows. A single colony of GG9 was inoculated in 2 mL Luria-Bertani (LB) medium and incubated overnight at 22 °C in an incubator shaker. The overnight cultures were 1:20 in 10 mL of fresh LB, and grown at 37 °C with shaking until mid-logarithmic phase (OD600 = 0.5). GG9 cells (5 µL aliquot) to be imaged were placed on the bottom of a glass bottom culture dish and covered with a 1% agarose pad made with LB supplemented with 250 µM (final concentration) of isopropyl β-D-1-thiogalactopyranoside (IPTG) to induce the expression of Sa-gpsB (Figure 2 and Video 2). Timelapse microscopy and cell length quantification were performed as described in the protocol section.
Inhibition of FtsZ
FtsZ, being a protein essential for cell division, is considered an attractive drug target and multiple groups are developing FtsZ inhibitors as a way to develop new antibiotics12. Localization patterns of FtsZ or one of the proteins associated with it, such as ZapA, can be used as a reporter to study and/or identify novel antimicrobial compounds. We use the protocol provided here to demonstrate this approach using S. aureus RB197 [SH1000 harboring pRB42 (PCd-zapASa-gfp bla erm)]9 and B. subtilis PE92 (ftsAZ::ftsAZ-gfpΩerm)13 strains. RB197 and PE92 strains were grown as described above in TSB (containing 5 µg/mL erythromycin; and 1.25 µM CdCl2 to induce the expression of zapA-gfp) and LB respectively. At mid-logarithmic phase, a well-characterized FtsZ inhibitor, PC19072314,15, was added at 2 µg/mL final concentration and its effect on the S. aureus and B. subtilis cells were monitored using microscopy at different time intervals (Figure 3 and Figure 4). Quantification of cell diameter of S. aureus and cell length of B. subtilis was performed as described in the protocol section.
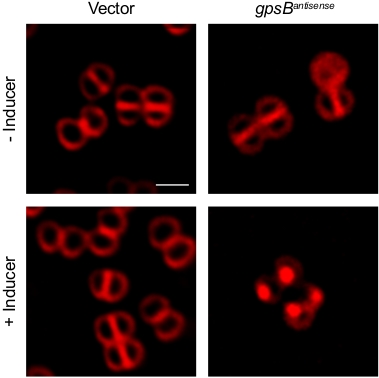
Figure 1: High-resolution micrograph of S. aureus cells displaying sick phenotype. Fluorescence micrographs of S. aureus strains harbouring either empty vector (left; RB143) or an inducible copy of antisense RNA of gpsBSa (right; GGS8) in the presence and absence of 1% xylose (inducer). Cells awere stained with FM4-64 membrane stain (stocks dissolved in sterile water) and imaged using TRITC filter set. Scale bar: 1 µm. Please click here to view a larger version of this figure.
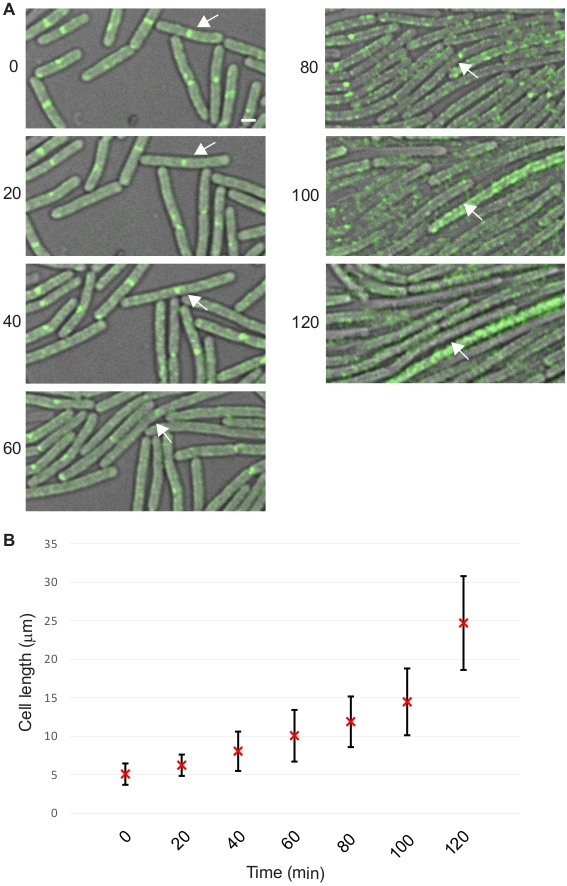
Figure 2: Representative data showing cell division inhibition in B. subtilis. (A) Timelapse micrographs of B. subtilis strain GG9 with images acquired at 20-min intervals for 120 min using the DIC/FITC channels. Fluorescence data of FtsZ-GFP (green) are shown. Arrows follow one cell throughout the experiment. Scale bar: 1 µm. (B) Quantification of cell lengths at all time points. Average cell length with error bars indicating standard deviation (n = 50) are shown. Please click here to view a larger version of this figure.
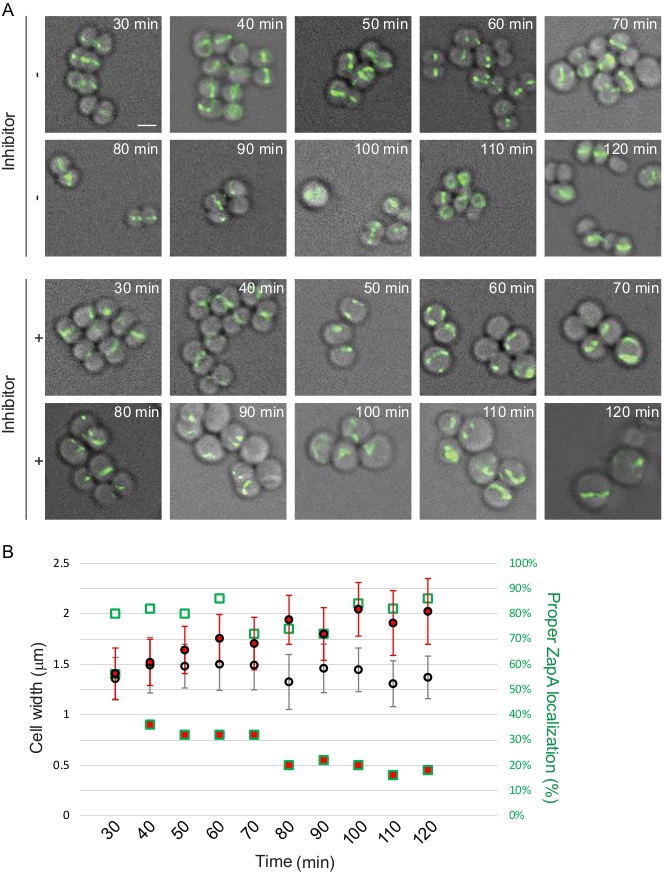
Figure 3: Time course investigation of cell division inhibition in S. aureus. (A) Mid-logarithmic phase cells of strain RB197 untreated (top) or treated (bottom) with FtsZ inhibitor (PC190723), subsequent to 30 min growth, aliquots of growing cultures were taken every 10 min for 90 min and imaged using DIC and FITC filter sets. Fluorescence from ZapA-GFP is shown. Scale bar: 1 µm. (B) Quantification of microscopy data. Average cell width with error bars indicating standard deviation (n = 50) and percentage of cells (n = 50) displaying proper ZapA-GFP localization (mid-cell and periphery) are shown. Data points of cells treated with inhibitor are shown in red. Shapes with green outline corresponds to the right Y-axis. Please click here to view a larger version of this figure.
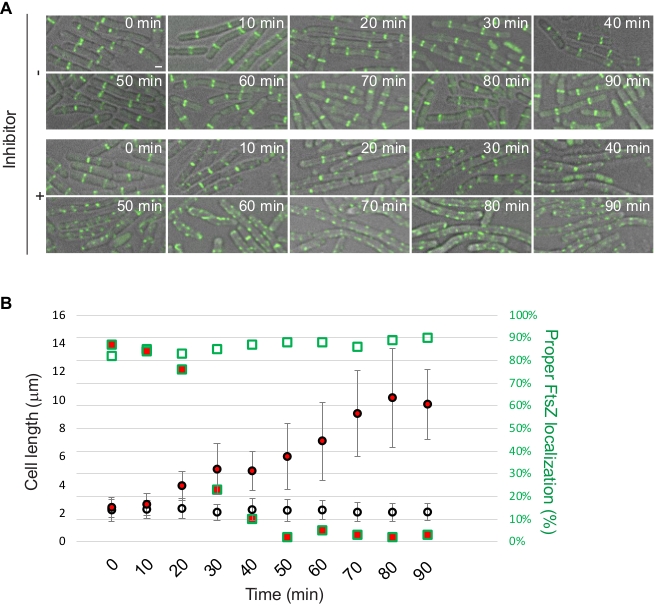
Figure 4: Investigation of cell division inhibition by a synthetic inhibitor in B. subtilis. B. subtilis strain PE92 was either untreated or treated with an FtsZ inhibitor (PC190723) at mid-logarithmic phase and were monitored for the subsequent 90 min. Aliquots of growing cultures were taken every 10 min for microscopy and images were acquired using DIC/FITC channels. Fluorescence from FtsZ-GFP is shown. Scale bar: 1 µm. (B) Quantification of microscopy data. Average cell length with error bars indicating standard deviation (n = 50) and percentage of cells (n = 50) displaying proper mid-cell FtsZ-GFP localization are shown. Data points of cells treated with inhibitor are shown in red. Shapes with green outline corresponds to the right Y-axis. Please click here to view a larger version of this figure.
Video 1: Timelapse microscopy of S. aureus cells developing sick phenotype. Strain GGS8 (gpsB antisense) treated with 1% xylose. Cells were stained with FM4-64 membrane stain and imaged at 10 min intervals for 60 min using the TRITC channel as described in the protocol. Please click here to download this video.
Video 2: Overexpression of Sa-gpsB leads to inhibition of cell division in B. subtilis. Timelapse video showing filamentation and change in FtsZ-GFP localization in GG9. Images were taken at 20 min intervals for 120 min using DIC and FITC channels. Please click here to download this video.
| Species | Strain | Genotype | Reference | |
| S. aureus | RB143 | SH1000 pEPSA5, bla, cat | Eswara et al, 2018 | |
| S. aureus | GGS8 | SH1000 pGG59 (pEPSA5 backbone) Pxyl-gpsBantisense, bla, cat | Eswara et al, 2018 | |
| S. aureus | RB197 | SH1000 pRB42 (pJB67 backbone) PCd-zapASA-gfp, bla, cat | Eswara et al, 2018 | |
| B. subtilis | GG9 | amyE::Phyperspank-gpsBSA spc; ftsAZΩftsAZ-gfp erm | Eswara et al, 2018 | |
| B. subtilis | PE92 | ftsAZ::ftsAZ-gfp Ωerm | Brzozowski et al, 2019 | |
Table 1: Strains used.
