This technique evaluated changes in mutants thought to affect the calcium handling or muscle depolarization. Baseline fluorescence levels and fluorescent transients were visualized and resting cytosolic calcium and calcium kinetics within the muscle were evaluated. It is important that the animals were grown on all-trans retinal for at least three days to ensure the successful incorporation of retinal, thereby subsequently activating the channelrhodopsin (Figure 2A). If animals are not exposed to all-trans retinal, no muscle calcium transient is triggered (Figure 2B). Although these animals can still be used to evaluate baseline cytosolic calcium levels, any dynamic changes in calcium will not be captured. Additionally, as an internal control for animal or dissection health, the animal will contract following blue light stimulation. A recording with a muscle contraction that causes minimal gross movement of the worm's body was selected for data collection when using nanobeads for immobilization. If the muscle used to collect the raw fluorescence values contracts vigorously, causing the whole body of the worm to move, the transient trace will reflect this motion artifact (Figure 2C) and should be discarded from quantification.
A mutation mapping to the sca-1 gene locus was isolated from a screen for mutations impacting C. elegans muscle nicotinic receptor localization. The C. elegans sca-1 gene encodes the only homolog of the sarco(endo)plasmic reticular calcium ATPase (SERCA) in the worm and is the only SERCA pump present in body wall muscles12,13. Loss-of-function sca-1 mutants were predicted to exhibit changes in the muscle calcium handling based on the important role SERCA plays in maintaining calcium homeostasis in mammalian muscles14,15,16,17,18. The GECI RCaMP was expressed in the body wall muscles and the blue light-activated channelrhodopsin was expressed in excitatory cholinergic neurons to evaluate both the intracellular baseline calcium and calcium dynamics in sca-1 mutants. With the dissected preparation technique, increases in baseline levels of RCaMP fluorescence were observed in the sca-1 mutant when compared to the control (Figure 3A,B), suggesting that loss of SERCA function leads to elevated levels of resting cytoplasmic calcium. When calcium dynamics were examined, following presynaptic stimulation triggered with a train of 5, 2 ms blue light pulses (Figure 3C), there was a significant decrease in peak calcium levels in the sca-1(tm5339) mutants as compared to the control (Figure 3D), which may reflect reduced calcium stores in the SR. However no change was seen in the rise to peak time or the half decay time (Figure 3E,F). This suggests that evoked changes in cytosolic calcium from either external source such as calcium entry through acetylcholine receptors and/or voltage-gated calcium channels as well as from internal stores are not affected in sca-1(tm5339) mutants. Similar results can be observed using the nanobead preparation, as shown in Figure 419. This recapitulation of data provides evidence that both of these techniques are physiological for measuring body wall muscle resting cytoplasmic calcium levels as well as observing stimulated calcium transients. This also demonstrates that the dissection of the worm does not cause damage to the animal that alters its calcium handling or the ability to depolarize postsynaptic muscles.
This protocol can also be used to examine mutants that indirectly impact calcium handling in C. elegans. Loss of function slo-1(eg142) mutants were evaluated to demonstrate this. The slo-1 gene encodes a calcium-activated BK potassium channel, which is expressed in both neurons and muscles20,21. Studies have previously described a role for SLO-1 in body wall muscles, demonstrating that loss of function mutants have a defect in postsynaptic repolarization following muscle action potentials20,21. Possible changes in baseline calcium levels and calcium transient dynamics due to this hyperexcitability were examined using nanobead immobilization. When baseline levels of RCaMP were measured, slo-1(eg142) mutants displayed increase fluorescence as compared to the controls (Figure 5A,B)19. This suggests that BK channels may also regulate baseline levels of cytosolic calcium. No changes in peak calcium levels were seen as compared to the control (Figure 5C,D) when evaluating the kinetics of the evoked calcium transient. The slo-1(eg142) mutants, however, exhibited a significantly increased rise to peak time as compared to the control (Figure 5E). This may reflect a previously reported increase in presynaptic excitability20,21. There was, however, no significant change in the half decay time in slo-1(eg142) mutants as compared to the control (Figure 5F). Together, these data demonstrate that this method can be used to evaluate the effects of pre- and postsynaptic mutations on both resting and stimulated body wall muscle calcium.
| Serial Setting | Value |
| Answer timeout | 500 |
| Baud rate | 57600 |
| DelayBetweenCharsMs | 0 |
| Handshaking | Off |
| Parity | None |
| StopBits | 1 |
| Verbose | 0 |
Table 1: Serial port settings for microcontroller plugin, which controls an external microcontroller board. This is used to program external timing pulses to control fluorescence excitation.
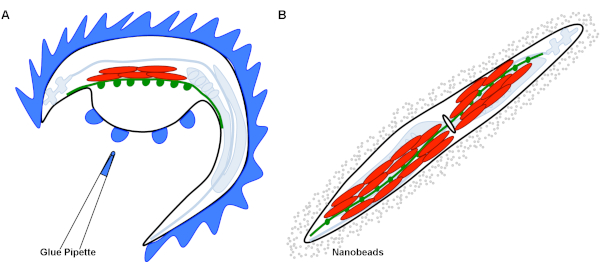
Figure 1: Graphical representation of different immobilization techniques. (A) This graphic demonstrates the dissection technique for immobilization. The body of the worm is glued down (blue) around the dorsal side of the animal. A dorsal incision along the cuticle generates a cuticle flap. This flap has been glued down to expose the intact NMJs formed between synapses of the ventral nerve cord in green (channelrhodopsin) and RCaMP expressing muscles in red. (B) This graphic demonstrates the nanobead immobilization technique. The intact worm is surrounded by a representation of the nanobeads in solution, that when compressed by the overlying coverslip, immobilize the worm. Due to the transparency of C. elegans, the ventral nerve cord can be visualized in green (channelrhodopsin) and RCaMP expressing muscles can be visualized in red. The ovoid in the middle of the worm represents the vulva, which is a landmark identifying the ventral body wall muscles. Please click here to view a larger version of this figure.

Figure 2: Representative traces of calcium transients. (A) Single trace representation of a calcium transient evoked by a train of 5, 2 ms blue light pulses at 50 ms interpulse intervals, recorded in an animal with limited body movement (- movement) grown in the presence of all-trans retinal (+ all-trans retinal). The spike artifacts are the blue light stimulation pulses, as both LEDs are co-illuminated simultaneously. (B) Single trace representation showing the absence of a calcium transient in response to a train of 5, 2 ms blue light pulses with 50 ms interpulse intervals in an animal that has not been exposed to all-trans retinal (- all-trans retinal, – movement). (C) Single trace representation of a calcium transient that has been evoked by a train of 5, 2 ms blue light pulses with 50 ms interpulse intervals, recorded in an animal that had significant muscle contraction leading to movement artifacts (+ movement, + all-trans retinal). Please click here to view a larger version of this figure.
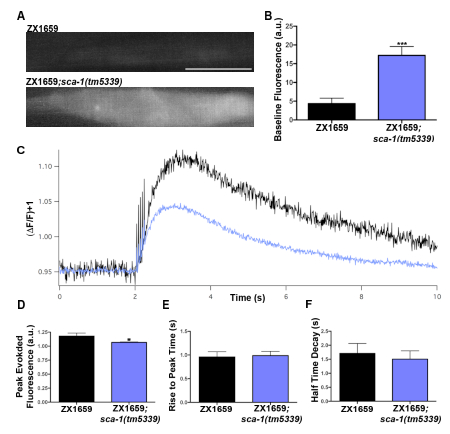
Figure 3: Baseline calcium levels and evoked calcium transients from control and sca-1(tm5339) samples prepared using the dissection technique. (A) Representative images of baseline RCaMP fluorescence in sca-1(tm5339) mutants and controls. Scale bar = 50 µm. (B) Baseline fluorescence levels quantification in sca-1(tm5339) mutants (n=13) and control (n=9). (C) Ensemble average traces of evoked calcium transients for sca-1(tm5339) mutants and control animals. (D) Quantification of peak RCaMP fluorescence from channelrhodopsin evoked calcium responses in sca-1(tm5339) mutants (n=12) and controls (n=9). (E) Quantification of the rise to peak time of channelrhodopsin evoked calcium transients in sca-1(tm5339) mutants (n=12) and controls (n=8). (F) Half decay time quantification of channelrhodopsin evoked calcium transients in sca-1(tm5339) mutants (n=12) and controls (n=7). Statistically significant values were: not significant p>0.05, * p≤0.05, ** p≤0.01, *** p≤0.001. Error bars represent mean ± SEM. Data were normalized using Shapiro-Wilk tests and significance was determined with a Mann-Whitney test for non-normal distributions. Please click here to view a larger version of this figure.
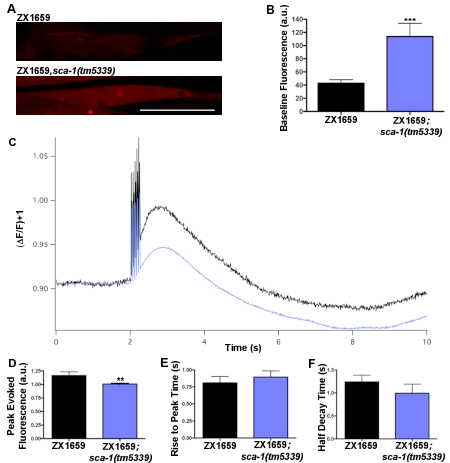
Figure 4: Baseline calcium levels and evoked calcium transients from control and sca-1(tm5339) samples prepared using nanobead immobilization. (A) Representative images of baseline fluorescence in sca-1(tm5339) mutants and controls, scale bar 50 µm. (B) Quantification of baseline RCaMP fluorescence levels in sca-1(tm5339) mutants (n=14) and the control (n=13). (C) Average of evoked calcium transients for sca-1(tm5339) mutants and controls. (D) Quantification of peak RCaMP fluorescence following evoked calcium response in sca-1(tm5339) mutants (n=14) and controls (n=13). (E) Quantification of rise to peak time of evoked calcium transients in sca-1(tm5339) mutants (n=14) and controls (n=12). (F) Quantification of half decay time of evoked calcium transients in sca-1(tm5339) mutants (n=14) and control (n=13). Figure is adapted from19. Statistically significant values were: not significant p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. Error bars show mean ± SEM. Data normality was assessed using Shapiro-Wilk tests and significance was determined with a Mann-Whitney test for non-normal distributions. This figure has been modified with permission from19. Please click here to view a larger version of this figure.
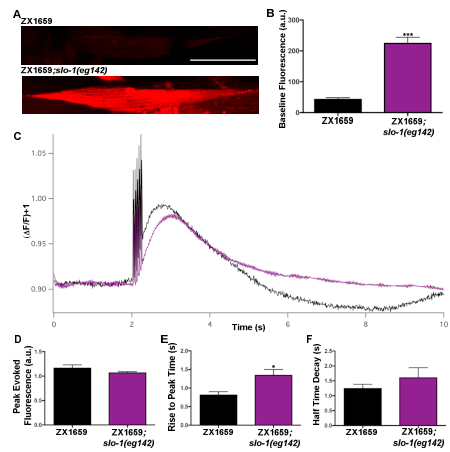
Figure 5: Baseline calcium levels and evoked calcium transients from control and slo-1(eg142) samples prepared using nanobeads immobilization. (A) Representative images of baseline fluorescence in slo-1(eg142) mutants and controls, scale bar 50 µm. (B) Quantification of baseline RCaMP fluorescence levels in slo-1(eg142) mutants (n=29) and the control (n=13). (C) Average of evoked calcium transients for slo-1(eg142) mutants and controls. (D) Quantification of peak RCaMP fluorescence following evoked calcium response in slo-1(eg142) mutants (n=29) and controls (n=13). (E) Quantification of rise to peak time of evoked calcium transients in slo-1(eg142) mutants (n=29) and controls (n=13). (F) Quantification of half decay time of evoked calcium transients in slo-1(eg142) mutants (n=11) and controls (n=13). Figure is adapted from19. Statistically significant values were: not significant p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. Error bars show mean ± SEM. Data normality was assessed using Shapiro-Wilk tests and significance was determined with a Mann-Whitney test for non-normal distributions. This figure has been modified with permission from19. Please click here to view a larger version of this figure.
