Mouse tendon fibroblasts were harvested from a previously described XMeCP2-LUC-HR/XMeCP2 female mouse, immortalized by retroviral transduction of E6 and E7 oncogenes from HPV-16 virus7, cloned using limiting dilution, and tested for luciferase activity and expression of wild-type MeCP2 protein (Figure 1A and 1B). Luciferase activity was robust if the reporter was on Xa, and undetectable if on Xi; the native MeCP2 expression exhibited a reciprocal pattern. We selected a clone with no luciferase activity (Xi-8) and a clone with robust activity (Xa-3) for further experiments. When Xi-8 cells were treated with 5-AZA, a dose-dependent induction in luciferase activity was observed (Figure 1C).
Sensitivity of Xi-8 and Xa-3 cells to hygromycin B was tested by performing 3 and 6 day-long selections. As expected from the expression pattern of the luciferase reporter, Xi-8 cells were more sensitive to hygromycin B than Xa-3 cells, even though the latter exhibited sensitivity to higher doses (Figure 2A). After a 1:100 mix of Xa-3 to Xi-8 cells was exposed to different concentrations of hygromycin B, a time-dependent increase in luciferase activity was observed, indicating successful competition of Xa-3 cells over Xi-8 cells (Figure 2B). Dose and duration of hygromycin B selection were chosen for further experiments based on preferential inhibition of Xi-8 over Xa-3 cells.
Four independent screens were performed to identify regulators of MeCP2 silencing using the Xi-8 cell line and an Open Biosystems miR-30- based shRNA library containing 64,159 different hairpins cloned into a MSCV-shRNA-pgk-Puro-ires-GFP retroviral vector. In each screen, twenty 10 cm plates were infected with pooled shRNAs, targeting approximately 100-fold coverage of the library. Hygromicin B was conducted for 6 days, until small acellular patches were observed; the cells continued to die for additional 24 to 48 hours. When recovered, the cells were harvested for DNA extraction using a phenol-chloroform technique. The total DNA yield per screen was around 20 µg/plate, which was pooled and divided between 100 PCR reactions using primers designed to amplify half-hairpins. The PCR products were then bead-purified and subjected to high throughput sequencing.
Pre- and post-selection samples were compared to determine hairpin enrichment, as we expected that any hairpin present at elevated levels in the post-selection sample targeted genes that reactivated Xi and thus conferred hygromycin resistance. Only hairpins enriched in at least 2 of the four screens were considered for validation. The most enriched hairpins in all replicates of our screen were four hairpins targeting XIST, consistent with its crucial role in XCI11.
The validation part of the screen was carried out by individually testing the hairpins that registered as hits for activation of the luciferase reporter in Xi-8 cells. Although the screen was carried out using an MSCV-based retroviral library, the validation was carried out using a lentiviral vector (individual pGIPZ clones with shRNA) to avoid possible detection of retrovirus-induced artifacts.
Each of the candidate genes was targeted by at least two different hairpin sequences, and the knockdown of the target gene was verified by real- time quantitative PCR (RT-qPCR). By assaying up to 1 x 106 cells in 6-well format and decreasing the background luminescence (Figure 2C), we were able to detect very low-frequency reactivation events, which we estimated to be in the 1:20,000 range.
Further analysis using individual hairpins led to the identification of 30 genes whose knockdown resulted in reactivation of the reporter on the Xi. The median magnitude of luminescence for these 30 genes was 2.8-fold above the baseline, which was further amplified to 7.1-fold following 5-AZA treatment. The same hairpins were retested, in conjunction with 5-AZA, with the Xa-3 cells to confirm re-expression of wild type MeCP2 from the Xi. This was achieved by performing RT-qPCR using primers designed to amplify only the wild type transcript of MeCP2.
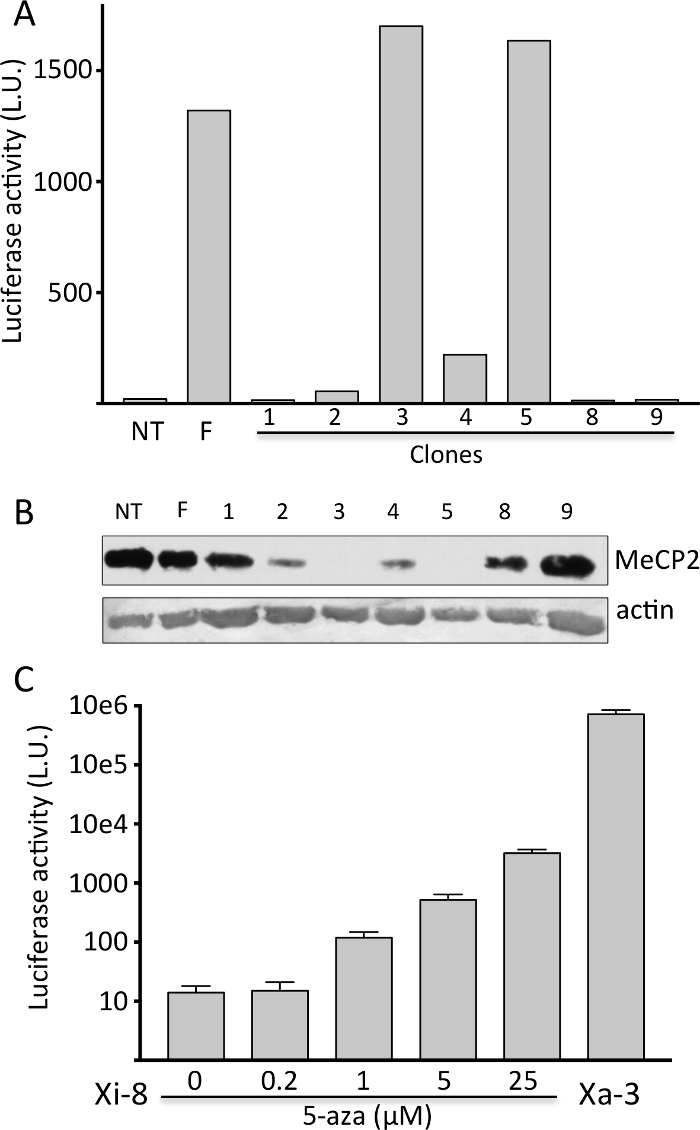
Figure 1. Isolation of clones with the MeCP2-Luc-HR reporter gene on the Xi.
A) Representative firefly luciferase assay on immortalized tendon fibroblasts clones (1-9). Primary fibroblasts from non-transgenic (NT) and transgenic heterozygous female (F) were used as negative and positive controls, respectively. B) Representative Western blot showing MeCP2 protein expression from the cells in (A). Note that luciferase activity (Figure 1A) and MeCP2 expression exhibit reciprocal expression pattern. C) Induction of firefly luciferase activity in Xi-8 cells with 5-AZA. Xa-3 cells, with the reporter gene on the Xa, serve as a positive control. Please click here to view a larger version of this figure.
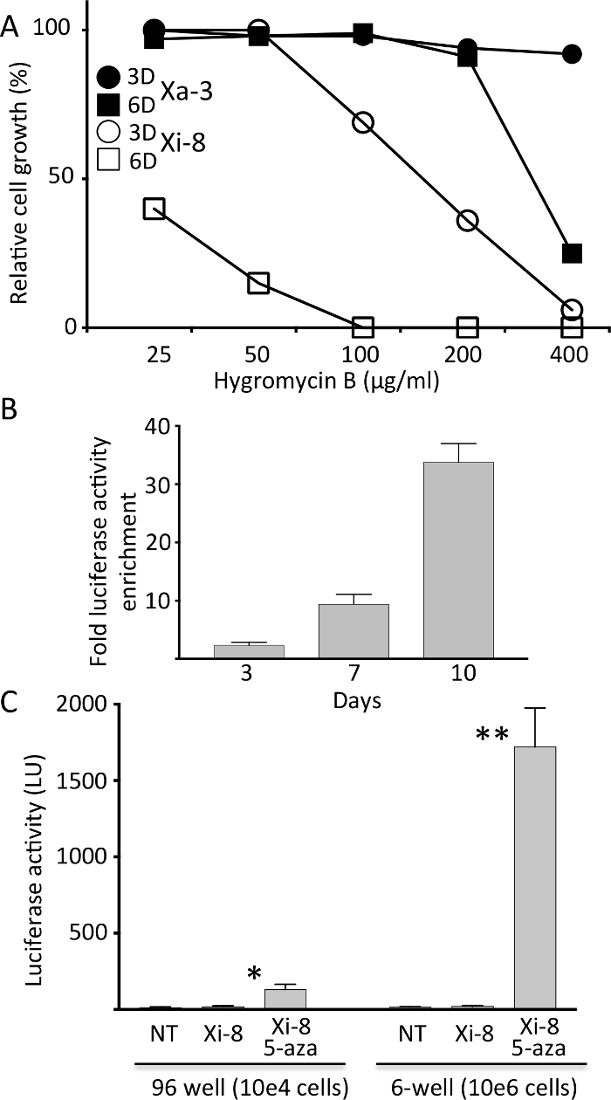
Figure 2. Characterization of the hygromycin resistance in Xi-8 and Xa-3 cells.
A) Relative growth of Xi-8 and Xa-3 cells treated with different hygromycin B regimens (3D = 3 days; 6D = 6 days), as compared to untreated cells. B) Luciferase activity enrichment after exposing a total of 10,000 Xa-3 and Xi-8 cells (mixed in 1:100 ratio) to hygromicin B (25 µg/mL) for indicated number of days. Post-exposure luciferase activity was divided by the pre-exposure level to calculate enrichment. C) Firefly luciferase activity in Xi-8 cells, assayed in 96- and 6-well format, with and without 5-AZA (10 µM). Non-transgenic (NT) fibroblasts were used as a negative control. 5-AZA increased the luciferase activity 7.2-and 86-fold over the baseline in 96- and 6-well format, respectively. (N = 3, *p < 0.001, **p < 0.0001 by Student's t- test comparing 5-AZA treated and untreated Xi-8 cells.) Please click here to view a larger version of this figure.
