Here we have described the process of measuring pHi in the follicle epithelium, which involves several steps. First, the ovaries are dissected from flies of the appropriate genotype using tools for dissecting and mounting (Figure 1). The ovarioles are then imaged using quantitative fluorescence microscopy and the images are analyzed to obtain measurements of pHi. For each image, the cell types of interest are identified as described in Section 3.1 (Figure 2). The ratio of the fluorescence intensities in the GFP and mCherry channels is converted to pHi values using a standard curve (Figure 3A) as described in Section 3.2. Using this method, we found that pHi increases with differentiation in the early FSC lineage, from 6.8 in FSCs, to 7.0 in prefollicle cells, to 7.3 in follicle cells (Figure 3B). The pHi values of each cell type can be represented graphically, as in Figure 3B, or as a pseudocolored micrograph that shows the differences in pHluorin to mCherry ratios (Figure 4 and Figure 5). In these images, differences in the pHluorin to mCherry ratios are displayed as differences in color, as defined by a LUT. It is important to select a LUT and maximum and minimum values for the range of colors to produce an image that is most representative of the data. Although, in general, the choice of LUT and range settings will not give the appearance of differences that are not present in the image, a less suited choice of LUTs may obscure pHi differences present in the micrograph (Figure 5).

Figure 1: Materials for dissection and mounting Drosophila ovarioles. (A) An image showing: (1) nail polish; (2) vacuum grease and beaker and pipet tip used for applying grease; (3) 23-gauge syringe needles; (4) Dumont Inox forceps, Size 5; (5) 3D printed mounting chamber; (6) 22 X 40 mM glass coverslips; (7) Round Glass Coverslips, 12 mm diameter, 0.13-0.16 mm thickness; and (8) 9-well glass dissecting dish. (B) A close-up image of the 3D printed mounting chamber. (C) An image showing a dissected pair of wildtype ovaries. (D) An image showing well-separated ovarioles. (E) A picture of the 3D chamber with dissected ovaries mounted under a round glass coverslip. The black dots are drops of nail polish used to hold the round coverslip in place. (F) An image of dissected ovarioles underneath a round glass coverslip after mounting. The black box indicates a region of the image with a single dissected ovariole. Scale bars represent approximately 500 µm. Please click here to view a larger version of this figure.
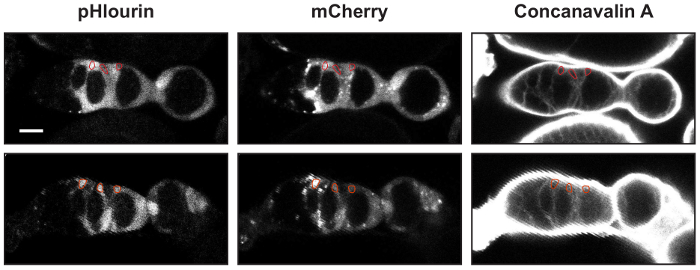
Figure 2: Identifying FSCs, pFCs, and follicle cells. Two examples of germaria with UAS-mCherry::pHluorin and 10930-Gal4 stained with Concanavilin-A. An ROI outlining an FSC, a pFC, and a follicle cell is shown in each image. The mean fluorescence intensities in the pHluorin and mCherry channels are used to calculate the pHluorin to mCherry ratios. Scale bars represent approximately 10 µm. Please click here to view a larger version of this figure.
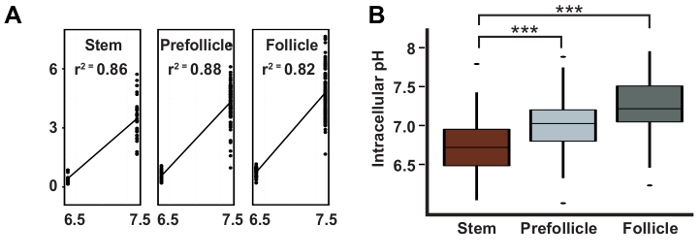
Figure 3: pHi increases during differentiation in the FSC lineage. Representative results taken from Ulmschneider et al.2 showing: (A) a typical linear regression curve used to calculate pH values from pHluorin to mCherry ratios; and (B) the calculated pHi values with 95% confidence intervals for FSCs, pFCs, and follicle cells. This figure has been adapted after permission, from Ulmschneider et al.2. Please click here to view a larger version of this figure.
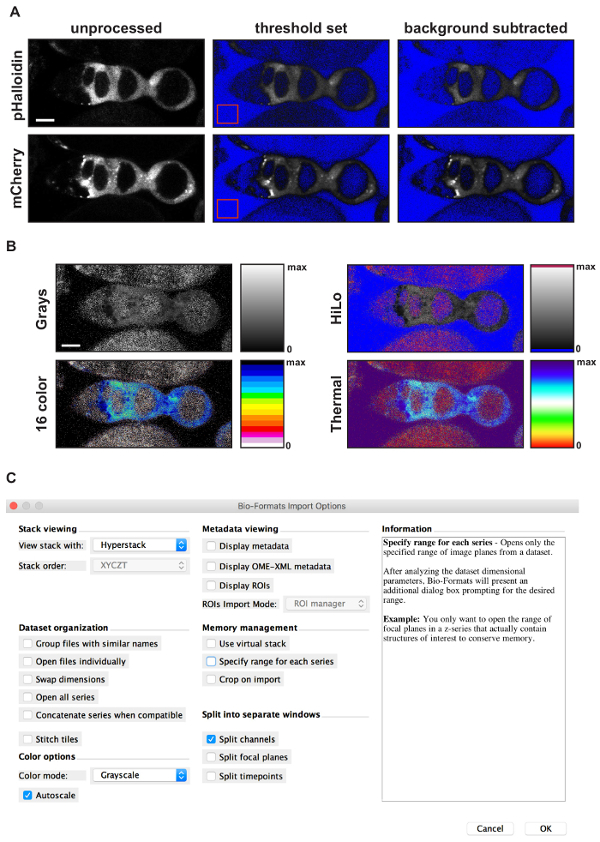
Figure 4: Using FIJI to generate a pseudocolored ratiometric image. (A) Images showing the results of each step in the background subtraction process. Starting with an unprocessed image (left panels), the first step is to set the threshold limits so that pixels with intensity values below background are excluded. The upper limit of the threshold is set to maximum. FIJI will color the excluded pixels blue, resulting in an image with a blue background and a clearly visible germarium (middle panels). Note, this step is optional. The second step is to draw an ROI in a part of the image without signal (red squares in middle panels), measure the mean fluorescence intensity of the ROI, and subtract that amount from the entire image, resulting in a background subtracted image (right panels). The third step is to use the Image calculation function to divide the image in the pHluorin channel by the image in the mCherry channel. The result will be an image displayed with the Greys lookup table and the image display values set to span the entire dynamic range provided by the bit depth of the image. The final step is to adjust the image display settings to a more appropriate range and select a lookup table. (B) Sample images showing the result of an image calculation with the minimum display value set to 0 and the maximum display value set to 2.5 using four different lookup tables. The rectangle next to each image shows the colors used across the dynamic range for each lookup table. Notice that for HiLo, 16 color, and Thermal, distinct colors are used for pixels with an intensity value of 0 or maximum (e.g., blue and red, respectively, in HiLo). This provides an easy visual reference of the limits of the dynamic range, allowing the viewer to see that the signal is within the dynamic range. (C) Screen shot showing the options selected when importing a file into ImageJ using the Bio-Formats Import Plug-In. Scale bars represent 10 µm. Please click here to view a larger version of this figure.
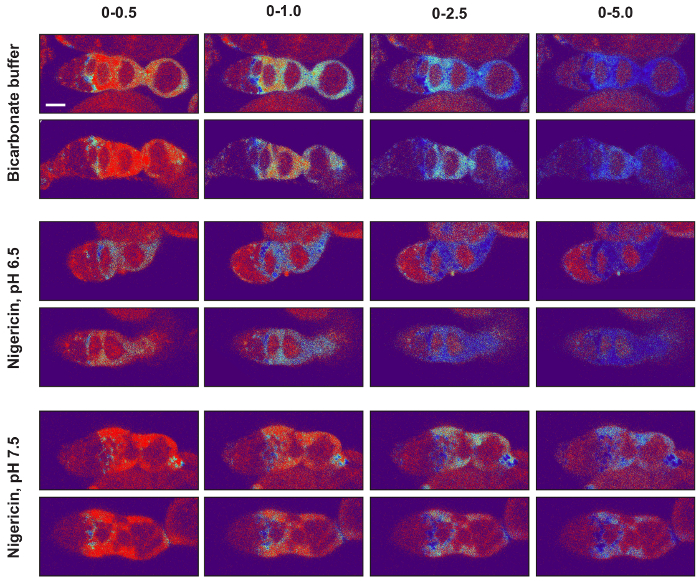
Figure 5: Setting the image display values of pseudocolored ratiometric images. The minimum and maximum image display values should be set so that the signal in images from the bicarbonate condition as well as from both nigericin conditions are within the dynamic range. To illustrate this point, two images from each of the three conditions are shown with four different image display settings (0-0.5, 0-1, 0-2.5, and 0-5) using the Thermal lookup table. Notice that, for all three experimental conditions, when the images are displayed with the maximum displayed value set to 0.5, much of the signal is at or near maximum on the colorimetric scale, and when it is set to 5.0, much of the signal is at or near minimum on the colorimetric scale. In both of these cases, the differences in the pHluorin to mCherry ratio across the tissue cannot be easily appreciated so these settings are not ideal. In contrast, maximum display values of 1.0 or 2.5 are much more appropriate. With these settings, the differences in ratios across the tissue can be easily appreciated, and the signals in the images from all three experimental conditions are displayed in colors that are within the dynamic range of the colorimetric scale. Scale bars represent approximately 10 µm. Please click here to view a larger version of this figure.
