The high throughput production of O2 probe pre-stained spheroids using micropatterned agarose method is schematically illustrated in Figure 1C showing examples of agarose microwells without and with the pre-stained spheroids. The efficiency of spheroid formation on agarose coating and their shape/sphericity can be cell-specific. For instance, human colon HCT116 cells never formed ideal spherical structures using agarose microwell method, in contrast to lipid-coated surface17, whereas hDPSCs alone and co-cultured with HUVEC always produced spheroids of highly reproducible shape and size proportional to cell composition, initial cell number, and duration of spheroid formation/growth. All the tested cell types efficiently accumulated O2 probe nanoparticles during spheroid formation (Figure 1B) and preserved this staining over at least 5 days, allowing their use for bioprinting and subsequent monitoring of the bioprinted tissue oxygenation (Figure 1D).
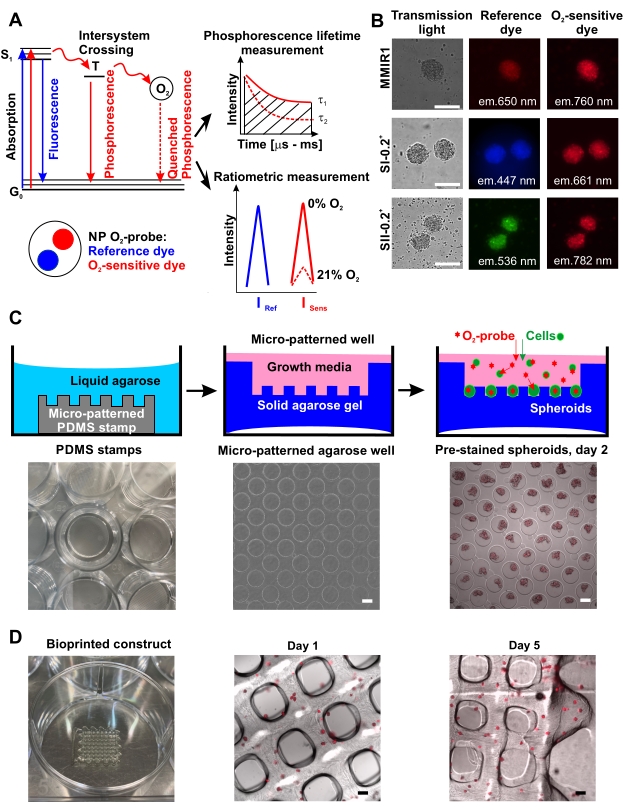
Figure 1: Principle of O2 probe function and its application for spheroid staining and bioprinting. (A) A simplified Yablonski diagram, explaining the principle of O2-sensing by the phosphorescent nanoparticle (NP) probes. The transfer of energy to molecular O2 during the forbidden triplet excited state (T) leads to a decrease of phosphorescence intensity inversely proportional to phosphorescence lifetime tau, τ (changes from τ1 at 0% O2 to τ2 at 21% O2), which is the time between excitation to singlet state (S1) and its return to the ground state (G0)42. Quantitative O2 measurement can be done by ratiometric measurement or by measuring τ (phosphorescence lifetime measurements).(B) An example of staining of human dental pulp stem cell (hDPSC) spheroids stained with different types of nanoparticle O2 probes, shown in transmission light, reference, and O2-sensitive dye fluorescence channels. Scale bar is 100 µm. (C) Schematics of high-throughput O2 probe pre-stained spheroid generation using micropatterned agarose method. From left to right: PDMS silicon stamp placed in a well of the culture plate, an example of a microwell pattern in agarose (4x magnification), and an example of MMIR1 pre-stained spheroids produced from HCT116 human colon cancer cells on day 2 after seeding on micropatterned agarose (4x magnification). Scale bar is 100 µm. (D) From left to right: a bioprinted waffle-construct and microscopy analysis of MMIR1 pre-stained spheroid bioprinted construct on days 1 and 5 after bioprinting. The fluorescence signal corresponds to O2-sensitive phosphorescent dye in MMIR1 nanoparticles (exc. = 635 nm/em. = 760 nm). Scale bar is 400 µm. Please click here to view a larger version of this figure.
On the described microscopy set-up (conventional widefield fluorescence microscope equipped with the light-emitting diode (LED) as a light source) red/near-infrared MMIR1 probe was found as the best in terms of photostability of its reference and sensitive dyes with the less than 5% decrease in brightness of their initial intensity ratio (R = Iref/Isens) after 12 cycles of continuous illumination. This allowed using MMIR1 probe for dynamic real-time study of rapid respiratory response in hDPSC spheroids upon treatment with mitochondrial uncoupler (FCCP) and inhibitor of complex I of electron-transport chain (rotenone) (Figure 2A,B). We found that in the stem cell-derived spheroids, FCCP displayed only mild uncoupling effect with a slight decrease of cell respiration within ~80 s after adding this drug, in agreement with previously reported metabolic features of DPSC40. On the other hand, rotenone strongly inhibited respiration leading to spheroids reoxygenation (reflected as the increase of Rp – spheroid periphery ratio and Rc – spheroid core ratio) and dissipation of the periphery-to-core O2 gradients within ~80 s after stimulation (Figure 2A). Two-point semi-calibration of MMIR1 probe ratio (R) at high (atmospheric) and low (in presence of sodium sulfite and glucose oxidase in imaging media) O2 levels confirmed the decrease of R, related to the decrease of oxygenation in spheroids with preliminary inhibited respiration by antimycin A/rotenone cocktail treatment (Figure 2C), illustrating how the measurement of MMIR1 probe R can be applied for semi-quantitative monitoring of long-term steady-state and quick oxygenation responses in 3D.
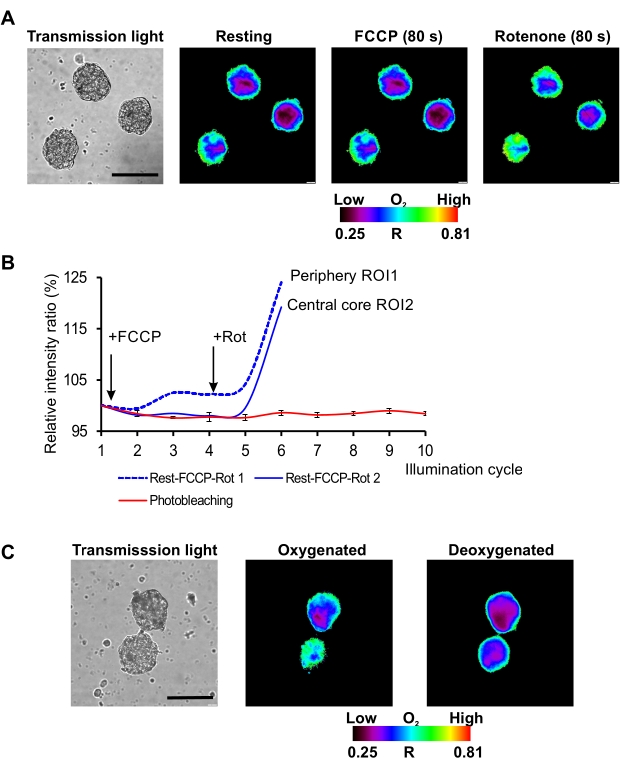
Figure 2: Kinetic analysis of O2 probe response to different stimuli. (A) Representative result of hDPSC spheroid oxygenation imaging at rest and in response to FCCP and rotenone treatments. Scale bar is 100 µm. (B) Kinetic response of MMIR1 intensity ratio to FCCP and rotenone treatment in comparison to the initial photobleaching intensity ratio kinetics (%).(C) Changes in the intensity ratio images of MMIR1 probe in hDPSC spheroids at oxygenated (antimycin A + rotenone addition) and deoxygenated (glucose oxidase addition) states.The color bar represents O2 probe intensity ratio (R = Iref / Isens) distribution across the image. Please click here to view a larger version of this figure.
To illustrate MMIR1 probe application for live metabolic imaging, the comparative analysis of O2 gradients in homocellular hDPSC versus heterocellular hDPSC/HUVEC (1:1) spheroids was performed. Taking advantage of the availability of free blue and green fluorescence spectral channels co-staining with Hoechst 34580 (HXT) and SYTOX Green (SYTOX) was performed to demonstrate the application of O2 probe for multiparametric studies (Figure 3). Using the automated protocol of pixel-by-pixel intensity ratio calculations provided by the imaging software false-color ratio images of R distribution in spheroids were produced, visualizing real-time detected periphery-to-core O2 gradients in all types of spheroids: with the oxygenated periphery and hypoxic niches in the center (Figure 2 and Figure 3A). However, Rp and Rc values, as well as the steepness of O2 gradient in the middle cross-sections varied for different spheroid types: see the line profiles of the middle cross-sections in hDPSC versus hDPSC/HUVEC spheroids and hDPSC versus bioprinted hDPSC spheroids (Figure 3B,C). As there was no widely accepted way of describing O2 gradients, we introduced several parameters allowing easy comparison and detailed description of general spheroid oxygenation: Rp and Rc, and such characteristics of O2 gradients as a difference between the oxygenated and hypoxic zones as (Rp-Rc), and average changes of oxygenation per µm as (Rp-Rc) / r, where r is a distance between periphery and hypoxic core, correlating in the middle cross-section with spheroid radius (Figure 3D). Statistical comparison of these parameters together with the data from necrotic dead cells visualized by SYTOX in spheroids, helped us to draw preliminary conclusions on the origin of the spheroid cell-type-specific O2 distribution gradients.
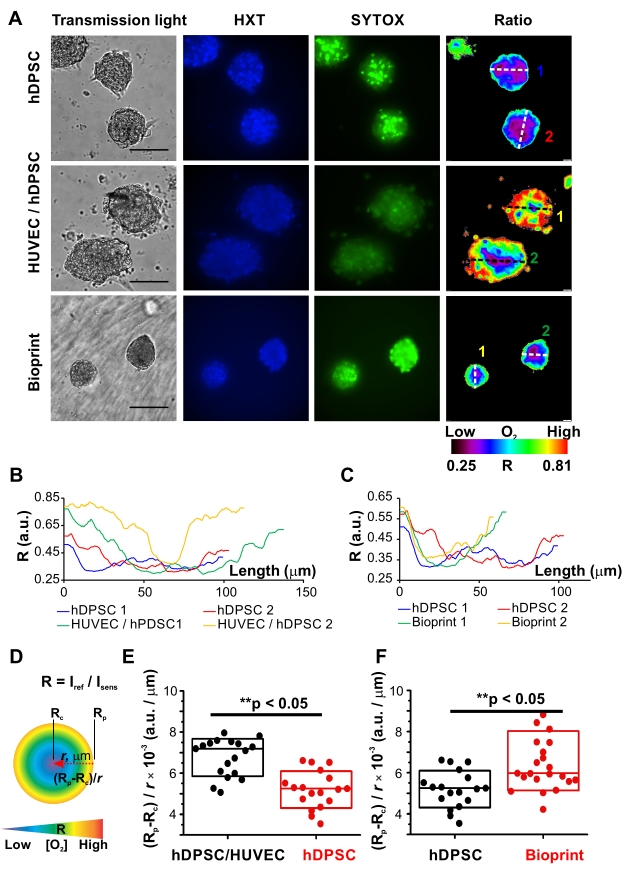
Figure 3: Comparative analysis of oxygenation gradients in spheroids. (A) Representative examples of live microscopy of oxygenation (ratiometric fluorescence microscopy of MMIR1) and live/dead cell staining (Hoechst 34580, HXT, blue and SYTOX Green, SYTOX) in homocellular hDPSC spheroids before and after printing, and heterocellular hDPSC/HUVEC (1:1) spheroids. hDPSCs in all types of spheroids were from the same cell culture. Spheroids were pre-stained with 5 µg/mL of MMIR1 probe for 2 days during their formation, and then used either for imaging or bioprinting (only hDPSC spheroids), with subsequent imaging analysis on day 1 after bioprinting. False-color images of the intensity ratio (Iref/Isens) images of MMIR1-stained spheroids correspond to their periphery-to-core O2 gradients. Scale bar is 100 µm. The color bar represents O2 probe intensity ratio (R = Iref/Isens) distribution across the image. Numbers 1 and 2 of diameter cross-sections correspond to intensity profiles shown in B and C. (B,C) Comparison of intensity ratio profiles between homogeneous hDPSC versus heterogeneous hDPSC/HUVEC spheroids (B) and homogeneous hDPSC spheroids before and after bioprinting (C). Profiles were measured for cross-sections of spheroids shown on (A). (D) Schematic representation of parameters used for the description of the periphery-to-core O2-gradients in spheroids, where r is a radius of spheroid and Rp and Rc are intensity ratios of the periphery and core region of spheroid correspondingly. The color bar schematically represents distribution of O2 gradient from high (red) to low (blue) levels in ideally spherical spheroid. (E,F) Comparative analysis of periphery-to-core O2-gradients (Rp-Rc) / r values in hDPSC/HUVEC and hDPSC spheroids (E) and in hDPSC spheroids before and on day 1 after bioprinting (F). Statistical analysis was done for one experimental repeat (n = 18-23). Boxes correspond to standard deviations. Asterisks indicate the statistical difference between groups (at p = 0.05); ** = p < 0.0005. Please click here to view a larger version of this figure.
In comparison to homocellular hDPSC spheroids, hDPSC/HUVEC spheroids had significantly steeper gradients: higher values of (Rp-Rc) / r parameter and range (Rp-Rc; Figure 3F). At the same time, they displayed higher oxygenation of the periphery (Rp) and core (Rc) (Figure 4A, Table 1). Interestingly, they were also statistically larger (Figure 4A, Table 1), suggesting that such differences in oxygenation were caused by their different cellular bioenergetic profiles. These data are in agreement with the well-known metabolic features of HUVEC cells having generally lower respiration activity of cells with the strong reliance on glycolysis and pentose-phosphate pathways as the main bioenergetic sources of ATP and NAD(P)H41. At the same time, the pronounced periphery-to-core O2 gradient formed in heterogeneous spheroids is likely generated by hDPSC in their composition, characterized by hyperpolarized mitochondria and active electron-transport chain40 and, according to the results on hDPSC spheroids oxygenation, having strong respiration activity (Figure 3, Figure 4A, and Table 1). Thus, generally higher viability of hDPSC/HUVEC spheroids confirmed by the lower intensity of their dead cells staining with SYTOX (Figure 3A) is potentially linked to their higher oxygenation levels.
To illustrate the applicability of live microscopy imaging of spheroid oxygenation in biofabrication, MMIR1 O2-probe pre-stained spheroids were used for bioprinting in GelMA bioink with the following comparison of O2 gradients in hDPSC spheroids before and on day 1 after bioprinting (Figure 3A,C,F, Figure 4B, and Table 2). Bioprinted hDPSC spheroids had significantly oxygenated periphery (higher Rp) in comparison to spheroids, measured before bioprinting, while their core oxygenation had similar values (Figure 4B). Changes in their periphery oxygenation affect the range (an increase of (Rp-Rc) and steepness (increased (Rp-Rc)/r) of their periphery-to-core O2 gradients, which were statistically different from hDPSC spheroids measured before bioprinting (Figure 3F and Figure 4B). The dead cells staining was generally brighter in bioprinted spheroids, suggesting that decreased spheroid viability is involved in the alteration of spheroid oxygenation (Figure 3A).
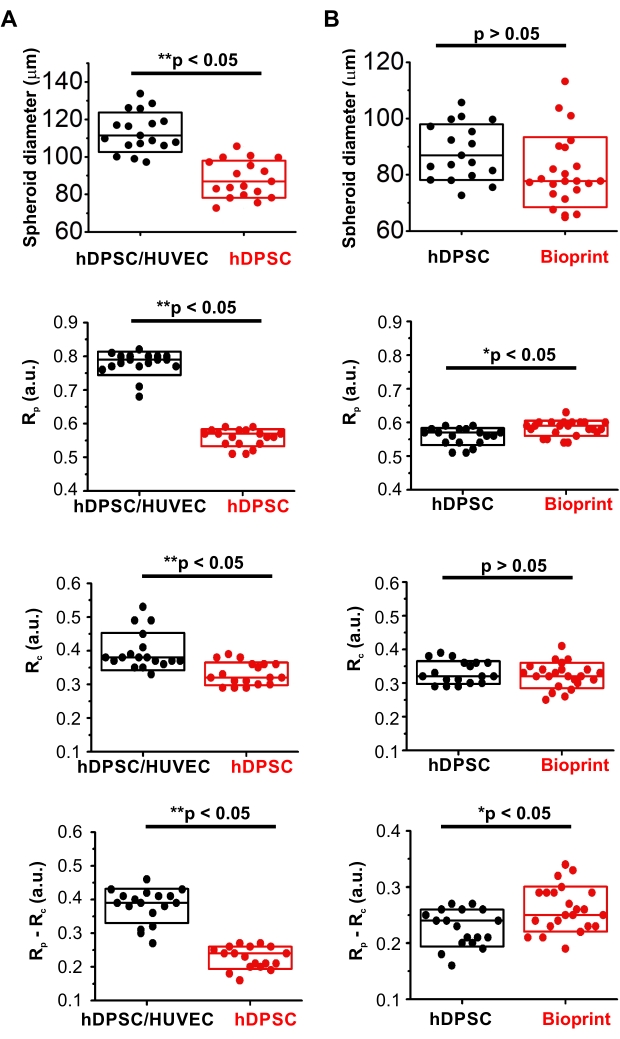
Figure 4: Comparative analysis of MMIR1-stained spheroids diameter and intensity ratio. (A) Comparison between hetero- hDPSC/HUVEC and homocellular hDPSC spheroids. (B) Comparison of homocellular hDPSC spheroids before and after bioprinting. Rp and Rc – intensity ratio at the periphery and the core of the spheroids correspondingly; (Rp-Rc) – the difference in intensity ratio, corresponding to the range of O2 gradient in spheroids. Statistical analysis was done for one experimental replicate (n = 18-23). Boxes correspond to standard deviations. Asterisks indicate the statistical difference between groups (at p = 0.05), where * = p < 0.005 and ** = p < 0.0005. Please click here to view a larger version of this figure.
| Type of spheroid (N) | Diameter [µm] | Rp [a.u.] | Rc [a.u.] | Rp-Rc [a.u.] | (Rp-Rc)/r [a.u./µm] |
| hDPSC / HUVEC (18) | 113.2 ± 10.6 | 0.779 ± 0.034 | 0.398 ± 0.055 | 0.982 ± 0.051 | 0.00677 ± 0.00091 |
| hDPSC (18) | 88.1 ± 9.9 | 0.558 ± 0.025 | 0.331 ± 0.034 | 0.227 ± 0.032 | 0.00520 ± 0.00090 |
| p-value | 1.5 x 10-8 * | 1.5 x 10-21 * | 1.3 x 10-4 * | 1.4 x 10-12 * | 9.2 x 10-6 * |
Table 1. Spheroid diameter, intensity ratio at the core (Rc) and periphery (Rp) of the spheroids, the difference in intensity ratio (Rp-Rc) and (Rp-Rc) / r value in heterogeneous hDPSC/HUVEC and homogeneous hDPSC spheroids. p-value of a t-test on N spheroids demonstrates statistical difference and is indicated with an asterisk as well.
| Type of spheroid (N) | Diameter [µm] | Rp [a.u.] | Rc [a.u.] | Rp-Rc [a.u.] | (Rp-Rc)/r [a.u./µm] |
| hDPSC (18) | 88.1 ± 9.9 | 0.558 ± 0.025 | 0.331 ± 0.034 | 0.227 ± 0.032 | 0.00520 ± 0.00090 |
| Bioprinted hDPSC (23) | 80.9 ± 12.5 | 0.584 ± 0.023 | 0.323 ± 0.038 | 0.261 ± 0.039 | 0.00658 ± 0.00144 |
| p-value | 0.054 | 0.0024 * | 0.46 | 9.9 x 10-4 * | 6.3 x 10-6 * |
Table 2. Spheroid diameter, intensity ratio at the core (Rc) and periphery (Rp) of the spheroids, the difference in intensity ratio (Rp-Rc) and (Rp-Rc) / r value in homogeneous hDPSC spheroids before and after bioprinting. p-value of statistical analysis (N spheroids). The statistical difference is indicated with an asterisk.
