Antibodies were conjugated to metal isotopes using commercially available labeling kits, according to the manufacturer's instructions. Antibody clones were validated by flow cytometry and mass cytometry prior to use in this panel. An initial list of clones was selected based on review of the literature and antibody availability. The expression levels of some ligands for NK cell receptors are low or undetectable on healthy PBMCs. Therefore, positive staining for some antibodies was validated by staining healthy PBMCs, chronic myeloid leukemia K562 cells, acute lymphoblastic leukemia NALM6 cells, or B cell acute lymphoblastic leukemia 697 cells (Supplemental Figure 1). Clones selected for the NK cell panel that did not produce an adequate stain or were too expensive were substituted for different ones, as detailed in Supplemental Table 1 and shown in Supplemental Figure 2.
Metal-isotope pairing with antibodies for these panels was performed using the principles outlined by Takahashi et al.16. Lineage markers were of medium to high intensity. Consequently, they were mainly conjugated to low and medium sensitivity masses leaving high sensitivity masses available for conjugation to antibodies against more dimly expressed markers. A publicly available panel designer software was used to detect abundance sensitivity (M ± 1 bleed) or oxidation (M + 16 bleed) issues and antibody-metal pairs were re-assigned accordingly. Additionally, several markers were conjugated on different metals with minimal differences noted on signal intensities (Supplemental Table 2 and Supplemental Figure 3). Antibody-metal pairings and clone information for the NK and ligand panels are listed in Table 1 and Table 2 respectively.
In-house conjugated antibodies were titrated on PBMCs at five different titers: 0.625, 1.25, 2.5, 5, and 10 µg/mL. The lowest antibody titer which resulted in the highest signal intensity and the best separation between positive and negative populations was selected based on visual assessment. Titrations for the NK and ligand panels are shown in Figure 1 and Figure 2 respectively. For certain markers, a clear distinction between positive and negative populations was not identified, due to the marker being dimly expressed or universally positive. To determine the most accurate working dilution for these antibodies, titers were assessed on multiple cell types (PBMCs, T cells, B cells, or NK cells), or on cell lines, to allow for identification of both positive and negative cell populations (Supplemental Figure 4). The staining index (SI) for each marker was not calculated as this metric is not applicable to CyTOF data17,18.
The panels described here were designed to be compatible with sample barcoding. There are several barcoding methods available for CyTOF. The most commonly used are a commercially available Palladium-based kit, which requires fixation prior to barcoding, and the CD45-based barcoding method described by Mei et al.15, which allows the barcoding of live cells. To assess which barcoding method best fit our needs, we tested the stability of NK cell marker staining after fixation in an early version of the NK cell panel (Supplemental Figure 5). We found that the expression of a majority of NK cell markers was affected by fixation. Consequently, we decided to use a modified two-of-four, CD45-based barcoding method on live cells15. This barcoding method uses 102Pd, 104Pd, 106Pd, and 108Pd, and differs from the three-of-six method originally described by Mei et al., which used 104Pd, 106Pd, 108Pd, 110Pd, 113In, and 115In. Indium channels were not included in our barcoding scheme, as they interfered with the signal from 115In-CD3. 110Pd was not included as it interfered with the signal from the HLA-DR Qdot and the CD19 Qdot in the NK cell and ligand panels, respectively.
Although we recommend NK cell purification prior to staining, the NK cell panel is designed to allow for the phenotyping of NK cells from whole PBMCs. An example of our NK cell gating strategy is shown in Figure 3A using PBMCs from a healthy donor. Staining and gates for each of the NK markers are shown on healthy, isolated NK cells in Figure 3B. The ligand panel is designed to detect the expression of NK cell ligands on whole PBMCs. Figure 4A illustrates the gating strategy used to identify CD4+ T cells, CD8+ T cells, NK cells, monocytes, and CD19+ B cells in PBMCs from a healthy donor. Representative staining examples for each ligand are shown in Figure 4B using PBMCs from acute dengue patients and HIV-infected individuals who were virologically suppressed.
To ensure panel stability over time, our protocol includes two possible options: lyophilization through a third-party company into single use beads or freezing of pre-made aliquots at -80 °C. For this protocol, the NK panel was lyophilized, and the ligand panel was frozen. Both methods were validated prior to using each panel on clinical samples.
We produced over 700 reactions of the NK panel from one master mix by performing multiple conjugations of each antibody in the panel. After validation and titration of each conjugated antibody, the antibodies were combined into a master mix, filtered through a sterile 0.1 µm syringe filter unit, and sent to a third party company for lyophilization. Two sets of single-stain lyospheres were made, one for surface staining, and one for intracellular staining. Antibodies not conjugated in-house (HLA-DR and CD16) could not be added to the lyosphere, due to the presence of antibody stabilizer, which interferes with the lyophilization process. These antibodies are added to the panel on the day of staining. A comparison between stains obtained pre- and post-lyophilization is shown in Supplemental Figure 6. The clone of LILRB1 antibody initially used in the lyospheres did not produce a sufficiently strong stain (Supplemental Table 1 and Supplemental Figure 2). A better clone was subsequently identified, conjugated and added to the panel on the day of staining (Table 1). The KIR2DS2 polyclonal antibody used in the lyopsheres was noted to produce a non-specific stain after lyophilization and we do not recommend its use for subsequent analyses (Supplemental Table 1 and Supplemental Figure 2). Most intracellular stains slightly increased in intensity following lyophilization (Supplemental Figure 6).
Prior to storage of the ligand panel master mix at -80 °C, we tested two different storage conditions. We prepared a smaller master mix of this panel and stored aliquots at -80 °C and in liquid nitrogen for approximately two months. After two months we stained whole PBMCs with the frozen aliquots. We compared the staining to that of PBMCs from the same donor stained with the freshly prepared panel (Supplemental Figure 7). We found storage at -80 °C and in liquid nitrogen does not change the signal intensity for most markers. In fact, the signal intensity of anti-pan HLA class I, anti-CD7, anti-CD4, anti-HLA-Bw4, anti-CD14, anti-CD11b, and anti-LFA-3 is higher upon freezing, particularly in the case of samples stored at -80 °C. We could not determine whether the signal intensities of anti-LLT-1, anti-Nectin-1, anti-MICA/B, anti-DR4/5, anti-ULBP-1,2,5,6, anti-Nectin-2, anti-CD155, and anti-B7-H6 were affected by freezing, due to the fact that healthy PBMCs do not express high levels of these markers. However, validation of these markers on cell lines (Supplemental Figure 1) was performed using conjugated antibodies stored at -80 °C. Consequently, we were confident that freezing did not result in a significant loss of signal. Signal intensity did decrease upon freezing for five markers: anti-CD8, anti-ICAM-1, anti-CCR2, anti-CD33, and anti-CD56. However, in all of these cases the clear separation between the positive and negative populations remained. Given that metal-conjugated antibodies are not stable at 4 °C for long periods of time, freezing was necessary to preserve panel stability long-term, and despite a decrease in staining intensity in a subset of markers, we were able to retain sufficient staining separation. Importantly, the loss of signal intensity of anti-CD8, anti-CCR2, and anti-CD56 was greater in the samples stored in liquid nitrogen compared to those stored at -80 °C. Based on this data, we decided to store the panel at -80 °C.
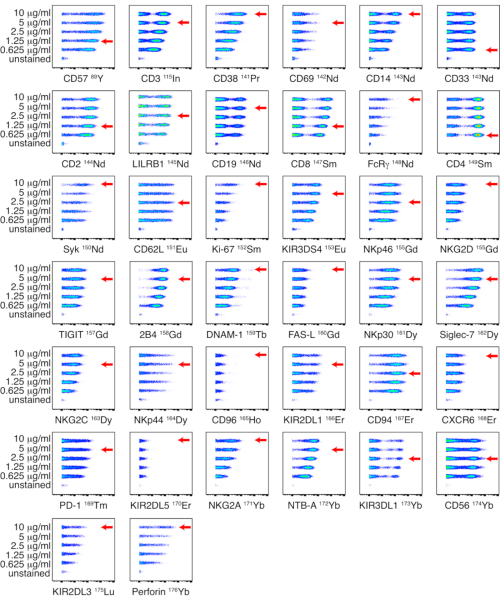
Figure 1: Titration of in-house conjugated antibody-metal conjugates for NK panel. Titrations of in-house conjugated antibodies were performed on PBMCs from a healthy donor using five different concentrations: 0.625, 1.25, 2.5, 5, and 10 µg/mL. Titers for anti-CD3, anti-CD14, anti-CD33, anti-CD19, anti-PD-1 and anti-CD56 were determined by gating on live cells. Titers for anti-CD4 and anti-CD8 were determined by gating on T cells. Titers for the remaining antibodies were determined by gating on NK cells. Since NKp44 is not expressed on resting NK cells, titers were determined on PBMCs stimulated with IL-2 and shown on NK cells. The red arrows indicate the titer selected for each antibody. Please click here to view a larger version of this figure.
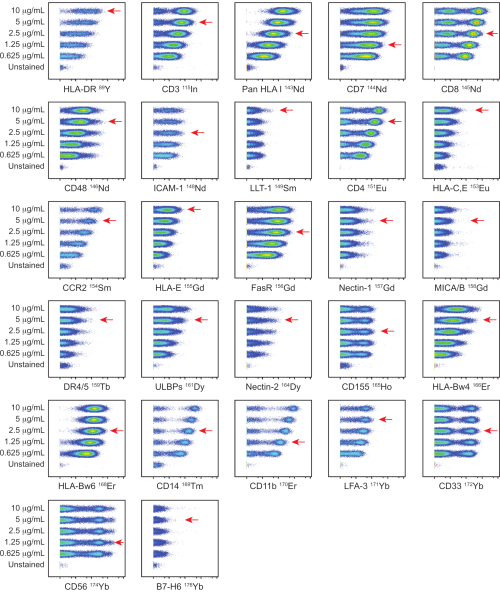
Figure 2: Titration of in-house conjugated ligand panel antibodies. Titrations of in-house conjugated antibodies were performed on PBMCs from a healthy donor using five different concentrations: 0.625, 1.25, 2.5, 5, and 10 µg/mL. Titers for anti-HLA-DR, anti-ICAM-1, anti-CCR2, anti-CD14, anti-CD11b, and anti-LFA-3 were determined by gating on CD3–CD7– cells. Titers for anti-CD3, anti-pan HLA class I, anti-CD7, anti-CD48, anti-LLT-1, anti-HLA-C,E, anti-HLA-E, anti-FasR, anti-Nectin-1, anti-MICA/MICB, anti-DR4/DR5, anti-ULBP-1,2,5,6, anti-Nectin-2, anti-CD155, anti-HLA-Bw4, anti-HLA-Bw6, anti-CD33, anti-CD56, and anti-B7-H6 were determined by gating on live cells. Titers for anti-CD4 and anti-CD8 were determined by gating on CD3+ cells. The red arrows indicate the titer selected for each antibody. Please click here to view a larger version of this figure.
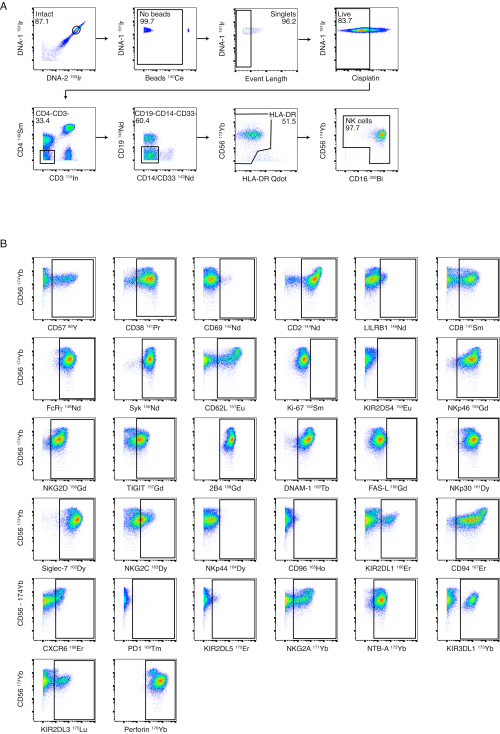
Figure 3: NK panel gating strategy and performance. (A) Serial negative gating from whole PBMCs to NK cells is shown in a healthy donor. Intact, bead and event-length gates ensure successful gating to single cells. Cisplatin staining was performed as a Live/Dead stain. T cells and B cells were excluded using CD3 and CD19. Monocytes were excluded by negative gating on CD4 and CD14/CD33 and by further negative gating of CD56-/HLA-DRbright cells. CD56 and CD16 were used to identify different subsets of NK cells (CD56bright, CD56dim and CD56–). (B) Examples of expression of NK cell receptors on NK cells from one healthy donor purified by magnetic-bead isolation. Please click here to view a larger version of this figure.

Figure 4: Ligand panel gating and performance. (A) Gating of major immune cell subsets from PBMCs derived from a healthy donor following normalization, calibration bead removal, and debarcoding. (B) Expression of ligands for NK cell receptors as well as several myeloid markers on live PBMCs. Staining for all ligands except Nectin-1 and B7-H6 is shown on PBMCs from acute dengue patients. Staining for Nectin-1 and B7-H6 is shown on PBMCs from HIV-infected individuals who were virologically suppressed. Please click here to view a larger version of this figure.
Supplemental Figure 1: Verification of antibody clones for ligand panel on cell lines. Antibodies against ligands for NK cell receptors that are expressed at low levels on healthy PBMCs were validated by staining cell lines. Chronic myeloid leukemia K562 cells were stained with anti-ICAM-1, anti-MICA/MICB, anti-DR4/DR5, anti-ULBP-1,2,5,6, anti-Nectin-2, anti-CD155, and anti-B7-H6. Acute lymphoblastic leukemia NALM6 cells were stained with anti-LLT-1 and B cell acute lymphoblastic leukemia 697 cells were stained with anti-Nectin-1. Dot plots and histograms showing staining on healthy PBMCs are in blue. Dot plots and histograms showing staining on the respective cell line are in red. The percentage of cells of the respective cell line that are positive for a given marker are provided. Please click here to download this file.
Supplemental Figure 2: Validation of antibody clones. (A) Different antibody clones were tested in healthy donors to identify the clone with the best specificity. 2B4, CXCR6, KIR2DS4, NKG2A and TIGIT are shown on NK cells. CD56 and LILRB1 are shown on live cells. (B) The KIR2DS2 antibody clone showed a non-specific stain after lyophilization. An example of staining on the same donor is provided pre- and post-lyophilization. Please click here to download this file.
Supplemental Figure 3: Optimization of antibody/metal pairs. Staining of PBMCs from healthy donors are shown. (A) Antibody/metal pairs tested for the panel. (B) Antibody/metal pairs used in the panel. LILRB1 and PD1 are shown on live cells. All the other markers are shown on NK cells. Please click here to download this file.
Supplemental Figure 4: Titration of dimly expressed and mostly positive NK cell markers. Titers for antibodies against NK cell markers that did not show a clear positive and negative population were assessed both on NK cells (red) and on either B cells (blue) or PBMCs (grey) from healthy donors. The arrows indicate the titer selected for each antibody. Please click here to download this file.
Supplemental Figure 5: Optimization of barcoding protocol. Epitope stability was tested before and after fixation with 2% paraformaldehyde on PBMCs from a healthy donor. (A) CD3, CD14 and CD56 staining was similar before (red) and after (blue) fixation, CD4 and CD16 staining was significantly affected by fixation. (B) Many NK cell markers were affected by fixation, including CD2, CD38, KIR3DL2, CD62L, KIR2DS4, NKp46, NKG2C, NKp30, NKG2D, KIR3DL1, TIGIT, KIR2DL1, KIR2DL3 and NTB-A. Please click here to download this file.
Supplemental Figure 6: Confirmation of panel stability after lyophilization. The stability of in-house antibody conjugates was confirmed by staining PBMCs from the same blood bank donor pre-lyophilization (blue) and post-lyophilization (red). Anti-CD3, anti-CD14, anti-CD33, anti-CD19, anti-PD-1, anti-CD56 stains are shown on live cells. Anti-CD4 and anti-CD8 are shown on CD3+ cells. Titers for the remaining antibodies are shown on NK cells, gated according to the gating scheme shown in Figure 1. Notably, anti-KIR2DS2 stained non-specifically after lyophilization and therefore has not been used for subsequent analyses. Please click here to download this file.
Supplemental Figure 7: Confirmation of ligand panel stability at -80 °C. The stability of in-house antibody conjugates was confirmed by staining healthy PBMCs from the same donor with a freshly prepared master mix as well as the same mix after storage at -80°C or in liquid nitrogen. Anti-HLA-DR, anti-ICAM-1, and anti-LFA-3 staining is shown on CD3–CD7– cells. Anti-CD3, anti-pan HLA class I, anti-CD7, anti-CD48, anti-LLT-1 anti-HLA-E, anti-FasR, anti-Nectin-1, anti-MICA/B, anti-DR4/5, anti-ULBP-1,2,5,6, anti-Nectin-2, anti-CD155, anti-HLA-Bw4, anti-HLA-Bw6, and anti-B7-H6 staining is shown on live cells. Anti-CD8 and anti-CD4 staining is shown on CD3+ cells. Anti-HLA-C,E, anti-CCR2, anti-CD11b, and anti-CD33 staining is shown on CD3–CD7–CD14+ cells. Anti-CD14 staining is shown on CD3–CD7–CD33+ cells and anti-CD56 staining is shown on CD3–CD7+CD14–HLA-DR– cells. Histograms showing staining with the freshly prepared panel are in red. Histograms showing staining with the panel after storage at -80°C for approximately two months are in blue. Histograms showing staining with the panel after storage in liquid nitrogen (LN2) for approximately two months are in green. Samples were stained and run on different days. Files were normalized and beads were removed using the premessa package. Please click here to download this file.
| Specificity | Clone | Isotope | Purpose | Surface/Intracellular |
| CD57 | HCD57 | 89Y | Maturity/Memory | surface |
| CD45 | HI30 | 102Pd, 104Pd, 106Pd, 108Pd | Barcoding | surface |
| HLA-DR | Tü36 | Qdot 655 (112Cd-114Cd) | Activation/Lineage | surface |
| CD3 | UCHT | 115In | T cell lineage | surface |
| CD38 | HIT2 | 141Pr | Activation Marker | surface |
| CD69 | FN50 | 142Nd | Activation Marker | surface |
| CD33 | WM53 | 143Nd | Monocyte lineage | surface |
| CD14 | M5E5 | 143Nd | Myeloid lineage | surface |
| CD2 | RPA-2.10 | 144Nd | Activation/Maturity | surface |
| LILRB1 | MAB20172 | 145Nd | Inhibitory Receptor | surface |
| CD19 | HIB19 | 146Nd | B cell lineage | surface |
| CD8 | SK1 | 147Sm | T cell lineage and NK cell Activation/Maturity | surface |
| FcRg | polyclonal | 148Nd | Maturity/Adaptive | intracellular |
| CD4 | SK3 | 149Sm | T cell lineage | surface |
| Syk | 4D10.2 | 150Nd | Signaling | intracellular |
| CD62L | DREG-56 | 151Eu | Activation | surface |
| ki-67 | Ki-67 | 152Sm | Proliferation | intracellular |
| KIR2DS4 | 179315 | 153Eu | Activating Receptor | surface |
| NKp46 | 9.00E+02 | 155Gd | Activating Receptor | surface |
| NKG2D | 1D11 | 156Gd | Activating Receptor | surface |
| TIGIT | 741182 | 157Gd | Inhibitory Receptor | surface |
| 2B4 | C1.7 | 158Gd | Activating Receptor | surface |
| DNAM-1 | DX11 | 159Tb | Activating Receptor | surface |
| FAS-L | NOK-1 | 160Gd | Apoptosis | surface |
| NKp30 | P30-15 | 161Dy | Activating Receptor | surface |
| Siglec-7 | S7.7 | 162Dy | Inhibitory Receptor | surface |
| NKG2C | 134522 | 163Dy | Maturity/Memory | surface |
| NKp44 | P44-8 | 164Dy | Activating Receptor | surface |
| CD96 | NK92.39 | 165Ho | NKG2 Co-receptor | surface |
| KIR2DL1/KIR2DS5 | 143211 | 166Er | Inhibitory Receptor | surface |
| CD94 | DX22 | 167Er | Activating Receptor | surface |
| CXCR6 | K041E5 | 168Er | Memory | surface |
| PD1 | EH12.2H7 | 169Tm | Inhibitory Receptor | surface |
| KIR2DL5 | UP-R1 | 170Er | Inhibitory Receptor | surface |
| NKG2A | 131411 | 171Yb | Inhibitory Receptor | surface |
| NTB-A | NT-7 | 172Tb | Activating Receptor | surface |
| KIR3DL1 | DX-9 | 173Yb | Inhibitory Receptor | surface |
| CD56 | NCAM16.2 | 174Yb | NK cell lineage | surface |
| KIR2DL3 | 180701 | 175Lu | Inhibitory Receptor | surface |
| Perforin | B-D48 | 176Yb | Cytolytic Protein | intracellular |
| DNA-1/DNA-2 | NA | 191Ir/193Ir | Nucleated cells | surface |
| Cisplatin | NA | 194Pt/195Pt | Viability | surface |
| CD16 | 3G8 | 209Bi | FcgRIII receptor | surface |
Table 1: NK panel. Markers are ordered according to the isotopic mass of the metal to which they were conjugated. 191Ir/193Ir is the natural abundance of the nucleic acid intercalator. 194Pt/195Pt is the natural abundance of cisplatin.
| Specificity | Clone | Isotope | Purpose | Surface/Intracellular |
| HLA-DR | L243 | 89Y | Antigen presenting cells, activation marker | surface |
| CD45 | HI30 | 102Pd, 104Pd, 106Pd, 108Pd | Barcoding | surface |
| CD19 | SJ25-C1 | Qdot 655 (112Cd-114Cd) | Lineage | surface |
| CD3 | UCHT1 | 115In | Lineage | surface |
| Pan HLA class I | W6/32 | 143Nd | KIR ligands | surface |
| CD7 | CD7-6B7 | 144Nd | Lineage | surface |
| CD8 | SK1 | 145Nd | Lineage | surface |
| CD48 | BJ40 | 146Nd | 2B4 ligand | surface |
| ICAM-1 | HA58 | 148Nd | LFA-1 ligand | surface |
| LLT-1 | 402659 | 149Sm | CD161 ligand | surface |
| CD4 | OKT4 | 151Eu | Lineage | surface |
| HLA-C,E | DT9 | 153Eu | KIR ligands | surface |
| CCR2 | K036C2 | 154Sm | Monocyte functional marker | surface |
| HLA-E | 3D12 | 155Gd | NKG2A/CD94 and NKG2C/CD94 ligand | surface |
| Fas (CD95) | DX2 | 156Gd | FasL receptor | surface |
| Nectin-1 | R1.302 | 157Gd | CD96 ligand | surface |
| MICA/B | 159227/236511 | 158Gd | NKG2D ligands | surface |
| DR4/5 | DJR1/DJR2-2 | 159Tb | TRAIL receptors | surface |
| ULBP-1/2,5,6 | 170818/165903 | 161Dy | NKG2D ligands | surface |
| Nectin-2 | TX31 | 164Dy | DNAM-1, TIGIT, and CD96 ligand | surface |
| CD155 | SKII.4 | 165Ho | DNAM-1, TIGIT, and CD96 ligand | surface |
| HLA-Bw4 | REA274 | 166Er | KIR3DL1 ligand | surface |
| HLA-Bw6 | REA143 | 168Er | KIR null allele | surface |
| CD14 | M5E2 | 169Tm | Lineage | surface |
| CD11b | ICRF44 | 170Er | Lineage | surface |
| LFA-3 | TS2/9 | 171Yb | CD2 ligand | surface |
| CD33 | WM53 | 172Yb | Lineage | surface |
| CD56 | NCAM16.2 | 174Yb | Lineage | surface |
| B7-H6 | 875001 | 176Yb | NKp30 ligand | surface |
| DNA-1/DNA-2 | NA | 191Ir/193Ir | Nucleated cells | surface |
| Cisplatin | NA | 194Pt/195Pt | Viability | surface |
| CD16 | 3G8 | 209Bi | Lineage | surface |
Table 2: Ligand panel. Markers are ordered according to the isotopic mass of the metal to which they were conjugated. 191Ir/193Ir is the natural abundance of the nucleic acid intercalator. 194Pt/195Pt is the natural abundance of cisplatin.
| Specificity | Clone | Vendor | Catalog Number | Surface/ Intracellular | Notes |
| 2B4 | 2-69 | BD Biosciences | 550814 | surface | new clone validated with improved staining |
| CD56 | N901 | Beckman Coulter | 6602705 | surface | new clone validated with improved staining |
| CXCR6 | 56811 | R&D Systems | MAB699 | surface | new clone validated with lower cost |
| KIR2DS2 | polyclonal | Abcam | ab175486 | surface | non specific staining noted after lyophilization – not used for analyses |
| KIR2DS4 | FES172 | Beckman Coulter | A60796 | surface | new clone validated with improved staining/lower cost |
| LILRB1 | GHI/75 | Biolegend | 333702 | surface | new clone validated with improved staining |
| NKG2A | Z199 | Beckman Coulter | IM2750 | surface | new clone validated with lower cost |
| TIGIT | MBSA43 | Thermo Fisher Scientific | 16-9500-82 | surface | new clone validated |
Supplemental Table 1: Antibodies for NK cell panel that were tested, but not used.
| Isotope | Specificity | Clone | Vendor | Catalog Number | Surface/ Intracellular |
| 143Nd | NKG2C | 134522 | R&D systems | MAB1381 | surface |
| 145Nd | CD38 | HIT2 | Biolegend | 303502 | surface |
| 146Nd | CD8 | SK1 | Biolegend | 344702 | surface |
| 149Sm | CD2 | RPA-2.10 | Biolegend | 300202 | surface |
| 151Eu | Siglec-7 | S7.7 | Biolegend | 347702 | surface |
| 154Sm | LILRB1 | 292319 | R&D systems | MAB20172 | surface |
| 163Dy | KIR3DL1 | DX-9 | BD biosciences | 555964 | surface |
| 168Er | CD62L | DREG-56 | Biolegend | 304802 | surface |
| 171Yb | PD1 | EH12.2H7 | Biolegend | 329902 | surface |
| 176Yb | CD69 | EH12.2H7 | Biolegend | 329902 | surface |
Supplemental Table 2: Antibodies for NK cell panel that were tested with a different antibody/metal pairing.
