After immunization of C57BL/6 mice, all mice were weighed, examined, and graded daily for neurological signs. The representative clinical course of EAE should result in a disease curve as presented in Figure 2A and a change of body weight in the mouse as presented in Figure 2B. C57BL/6 mice immunized with MOG35-55 usually started to develop disease symptoms around day 10–12 and achieved the peak of disease around day 15–21 after active immunization (Figure 2A). Weight change was a valuable indicator in the EAE model. Before the onset of disease, the body weight of immunized mice gradually increased, then typically decreased correlating to the increasing disease symptoms. At the peak of EAE, mice also showed the lowest body weight (Figure 2B). Then body weight recovered slightly as clinical symptoms decreased. However, the mice usually did not fully recover. C57BL/6 mice developed a monophasic chronic disease pathology upon MOG35–55 challenge (Figure 2A).
The clinical severity of EAE is directly associated with autoreactive T cell activation14. Neuroantigen-specific CD4+ T cells are capable of initiating and sustaining neuroinflammation and pathology in EAE3. The typical characteristics of CD4+T cells in the peak of EAE are shown in Figure 3. Further, the proportion of Th1 and Th17 subsets in encephalitogenic cells, which are the major pathogenic cells mediating EAE, was analyzed by flow cytometry. Following the separation of a single-cell suspension from brain, the cells were stained with CD45, CD11b, CD3, CD4, IFN-γ, and IL-17 antibodies, which are expressed by T lymphocytes. FSC-A vs. FSC-H and SSC-A vs. SSC-H were used to gate singlets (Figure 3A, B), then FSC-A vs. SSC-A were used to gate live cells based on size and granularity (Figure 3C). After, CD45+ CD11b– cells were gated to identify leukocytes excluding monocytes (Figure 3D). Then, the gate was set on CD3+CD4+ to identify CD4+ T lymphocytes (Figure 3E). Finally, IFN-γ and IL-17 were used to identify Th1 and Th17 subsets and assess the functional effect according to effector cytokines (Figure 3F). According to the clinical severity of disease, representative results showed that IFN-γ–producing Th1 and IL-17–producing Th17 cells significantly increased in the encephalitogenic cells from EAE mice.
| Grade | Clinical sign |
| 0 | no disease |
| 1 | decreased tail tone or slightly clumsy gait |
| 2 | tail atony and moderately clumsy gait and/or poor righting ability |
| 3 | limb weakness |
| 4 | limb paralysis |
| 5 | moribund state. |
Table 1: Clinical scoring system. C57BL/6 mice were immunized with the MOG35–55 peptide. Then, neurological signs were recorded. A 5-point scoring system was used to assess the severity of EAE.
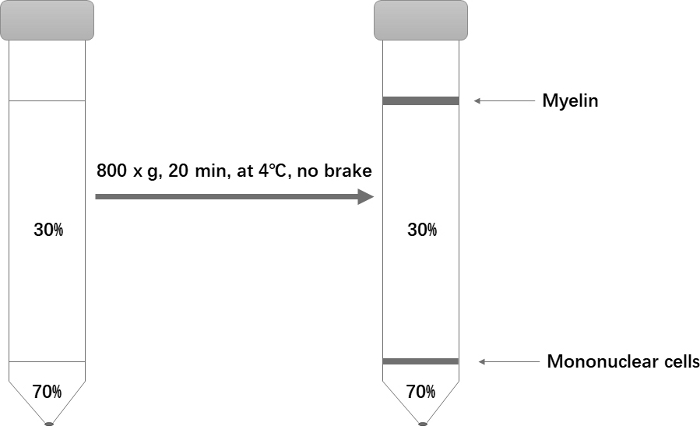
Figure 1: Schematic of the Percoll gradient setup for isolation of mononuclear cells. Please click here to view a larger version of this figure.
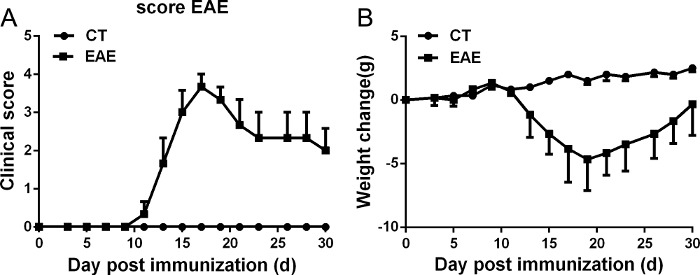
Figure 2: Representative course of EAE. EAE was induced in C57BL/6 mice by injection of MOG35–55 as described in the protocol. The clinical score (A) and change of body weight (B) were determined in these mice. Data are presented as mean ± SEM; n = 8 for each group. Please click here to view a larger version of this figure.
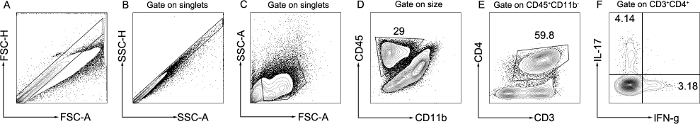
Figure 3: Representative flow cytometry analysis of lymphocytes in brain. A single-cell suspension was isolated from the brain in the peak of EAE. The gating strategy of T lymphocytes is shown. Singlets were gated as FSC-A vs. FSC-H and SSC-A vs. SSC-H (A,B). Live cells were gated as FSC-A vs. SSC-A (C). Leukocytes excluding monocytes were gated as CD45+ CD11b– (D). CD4+ T lymphocytes were gated as CD3+CD4+ (E). Th1 and Th17 subsets were gated as IFN-γ+ and IL-17+ (F). Please click here to view a larger version of this figure.
