Cómo estabilizar proteínas: pantallas estabilidad térmica cambio ensayos y diferencial de Nano fluorimetría en el proyecto de Virus X
Summary
Un protocolo se presenta para probar rápidamente la estabilidad térmica de las proteínas en una variedad de condiciones a través de ensayos de cambio térmico y diferencial fluorimetría de nano. Los sistemas tampón, sales y aditivos, juntos con tres pantallas de estabilidad única, se analizan con proteínas para identificar los tampones adecuados para estudios funcionales y estructurales.
Abstract
El proyecto Horizon2020 Virus-X se estableció en 2015 a la virosphere de biotopos extremos seleccionados de explorar y descubrir nuevas proteínas virales. Para evaluar el valor potencial biotecnológico de estas proteínas, el análisis de la proteína las estructuras y funciones es un desafío central en este programa. La estabilidad de la muestra de proteína es esencial para proporcionar resultados significativos y aumentar el crystallizability de los objetivos. El ensayo de cambio térmico (TSA), una técnica basada en la fluorescencia, se establece como un método popular para la optimización de las condiciones para la estabilidad de la proteína de alto rendimiento. En TSA, los fluoróforos empleadas son tintes extrínsecos, ambientalmente sensible. Una alternativa técnica similar es nano diferencial fluorimetría (nanoDSF), que se basa en la fluorescencia nativa de la proteína. Presentamos aquí una novela osmolyte pantalla, una pantalla de 96-condición de aditivos orgánicos diseñados para ensayos de cristalización a través de experimentos preliminares de TSA. Junto con pantallas sal y pH desarrollado previamente, el conjunto de tres pantallas ofrece un exhaustivo análisis de estabilidad de la proteína en una amplia gama de aditivos y sistemas de amortiguación. La utilidad de las pantallas se demuestra en el análisis TSA y nanoDSF de la lisozima y la proteína X, una proteína de la blanco del proyecto Virus X.
Introduction
Muchas enzimas útiles biotechnologically originan de fuentes virales, tales como el tabaco etch virus (TEV) proteasa1 y rinovirus humano tipo 3 C (HRV 3C) proteasa2. El proyecto (figura 1) Horizon2020 Virus-X3. El objetivo de este programa es (a) extender el rango de las propiedades de las familias de la enzima conocida y (b) caracterizar nuevas enzimas de función aún desconocida (enzima X). Determinación de la estructura cristalográfica desempeña un papel fundamental en la caracterización de la proteína objetivo, particularmente en aquellos casos en donde las secuencias de proteínas han evolucionado más allá de reconocimiento4. Estabilidad de la proteína es un factor clave en el proceso de cristalización; las muestras deben ser conformationally homogénea y estructuralmente durante un período de tiempo a forma de alta calidad, cristales de diffracting. Además, es esencial para los ensayos de actividad que las proteínas existen en su conformación activa, que también puede ser facilitado por un favorable entorno molecular.
A pesar del desarrollo en la tecnología disponible para cristalógrafos, cristalización de proteínas sigue siendo un proceso largo y requiere mano de obra empírica5. Experimentos preliminares biofísicos para mejorar la estabilidad de la proteína en solución dan claramente un punto de partida mejor para la cristalización de proteínas y generalmente consuman sólo una cantidad comparativamente pequeña de proteína muestra6,7, 8,9. La gran cantidad de proteínas de la blanco que se estudiarán en este proyecto también requiere un análisis de estabilidad, escalable, de alto rendimiento. Uno de los métodos más populares para la caracterización biofísica de la cristalización de las proteínas es el ensayo de cambio térmico (también conocido como TSA o diferencial de fluorimetría, DSF)10,11.
TSAs emplean un colorante fluorescente ambientalmente sensible a la desnaturalización térmica de las muestras de proteína. Muchos colorantes utilizados comúnmente tienen actividad de fluorescencia variable dependiendo de la polaridad de su entorno, a menudo mostrando una salida de alta fluorescencia en entornos hidrófobos pero sometidos a enfriamiento rápido en ambientes polares12. Proteínas generalmente causan aumentos pronunciados en la fluorescencia del tinte como sus corazones hidrofóbicos verse expuestos durante la desnaturalización, a menudo seguida por una disminución de la fluorescencia del tinte a muy altas temperaturas como las proteínas comienzan a agregados (figura 2).
Mientras que un tinte sensible a la hidrofobicidad es a menudo una buena opción para un tinte TSA de uso general, puede ser inadecuado para proteínas con regiones hidrofóbicas grandes, expuestos al solvente, que a menudo muestran fluorescencia del fondo perjudicialmente altos. Fluoróforos con modos alternativos de acción existen (ver discusión), pero en cambio puede ser deseable para seguir la desnaturalización a través de la fluorescencia intrínseca de proteínas con nanoDSF.
Residuos de triptófano que están enterrados en las regiones no polares de una proteína es fluorescente con un máximo de emisión de 330 nm. Como una muestra de la proteína se despliega y estos residuos se convierten en expuesto a un solvente polar, su máximo de emisión somete a un cambio bathochromic a 350 nm13. nanoDSF aprovecha este cambio de emisión máxima para el despliegue de una muestra de proteína sin necesidad de fluoróforos extrínsecos14.
Derretir las curvas que muestra pasos de desnaturalización solo pueden analizarse introduciendo datos a un modelo sigmoidal de Boltzmann. La temperatura en el punto de inflexión de la transición de despliegue (Tm) se utiliza como una medida cuantitativa de estabilidad térmica de la proteína y un punto de referencia para comparar la favorabilidad de diversas condiciones.
Curvas de fusión de la misma proteína en diferentes condiciones a veces poseen un grado de heterogeneidad que puede hacer inviable un ajuste sigmoidal de Boltzmann. Para discernir los valores dem de T de los datos que se desvía de la topología clásica curva, pueden utilizarse métodos numéricos tales como los empleados en NAMI, un programa open source TSA datos análisis11. Marcos termodinámicos alternativos también pueden utilizarse para analizar curvas más complejas con múltiples pasos de desnaturalización, como la metodología de ProteoPlex15.
Las pantallas de estabilidad fueron diseñadas para su uso en experimentos de TSA y nanoDSF para identificar rápidamente las condiciones favorables para una proteína diana (figura 3, composiciones de pantalla están disponibles en la información complementaria). Información recogida con las pantallas se puede utilizar en muchas etapas de la pipeline cristalográfico, incluyendo: muestra de almacenamiento; purificación, minimizando la pérdida de rendimiento a través de la proteína desplegar durante el proceso de purificación; ensayo diseño, reforzar la funcionalidad de la proteína en los ensayos de actividad con estabilización térmica almacenadores intermediarios y finalmente la cristalización, guía ensayos de cristalización racionalmente diseñado.
Elegir una base de sistema tampón adecuado para una muestra de la proteína es vital; valores de pH incompatibles pueden conducir a la desactivación o la desnaturalización de una proteína. Sin embargo, la presencia de moléculas de co cristalizado búfer resuelto en un gran número de estructuras cristalinas de la radiografía (tabla 1) también podría ser indicativa de un efecto estabilizador que está separado a la regulación de pH simple y en lugar de ello se deriva la química características de la molécula de búfer.
Formulado utilizando varios buffers17,18,19 junto a otros sistemas comúnmente biológicamente compatible con buffer de buenos, la pantalla de pH está diseñada para deconvolucionar el efecto químico de una molécula de tampón en estabilidad de la proteína desde el pH real de la solución resultante. Proporciona tres valores de pH para cada sistema buffer e incorporando redundancia del valor de pH entre los diferentes sistemas, la pantalla de pH puede identificar valores favorables de pH y sistemas buffer favorable para una proteína de la blanco.
La pantalla de sal contiene comúnmente sales de laboratorio así como chaotropes, chelants, metales pesados y agentes reductores. La pantalla puede dar una indicación general de la afinidad de una muestra de proteína a los entornos con alta fuerza iónica, pero cada subgrupo de los compuestos también puede proporcionar información sobre la posible estructura de una proteína. Por ejemplo, un fertilizantes quelatados significativamente desestabilizar una proteína podrían ser indicativo de metales estructurales importantes dentro de la muestra. Si la muestra es también fuertemente estabilizada por un catión metálico dentro de la pantalla, esto puede proporcionar un punto de partida prometedor para experimentos estructurales adicionales.
Osmolitos son compuestos solubles que afectan las propiedades osmóticas de su medio ambiente. En la naturaleza, pueden ser utilizados como “chaperonas químicas”, aplicar el plegamiento de proteínas desordenadas y estabilización, especialmente en condiciones de estrés20,21,22. Estas características hacen atractivas aditivos en Cristalografía de la proteína; utilizable como crioprotectores durante la cosecha de cristal, montaje y almacenamiento procesos23. Uso potencial de osmolitos se extiende también a la purificación de proteínas. Una proporción significativa de las proteínas recombinantes expresadas en e. coli puede ser insoluble y difícil de recuperar en el estado nativo usando métodos de purificación estándar. Osmolitos pueden utilizarse para estabilizar y salvar fracciones insolubles de proteínas, purificación cada vez mayor rinde24.
La pantalla del osmolyte fue diseñada usando establecidos compuestos presentes en el Protein Data Bank entradas25 y la base de datos de Dragon Explorer of Osmoprotection-Associated vías (DEOP)26 y había optimizado iterativamente usando proteínas estándar. La pantalla está construida alrededor de ocho subclases de osmolyte: glicerol, azúcares y polioles, detergente no sulfobetaines (NDSBs), betaínas y sus análogos, organofosforados, dipéptidos, aminoácidos y sus derivados y un último grupo misceláneo. Cada osmolyte está presente en concentraciones múltiples basadas en su solubilidad y rangos de concentración efectiva para comparación.
Protocol
Representative Results
Discussion
Los aspectos más importantes dentro del protocolo incluyen el paso de centrifugación y correcto sellado de la placa de 96 pocillos para experimentos TSA (criterio 1.5). Centrifugación asegura de que la condición de la pantalla y muestra de proteínas entran en contacto y mezcla. Además, si se utiliza una placa sin sellar para un experimento TSA, existe un riesgo significativo de solventes de evaporación durante todo el experimento, causando un aumento en la concentración de la muestra y aumenta las posibilidades de agregación de la proteína.
TSAs y nanoDSF son susceptibles a una amplia gama de muestras de proteínas; la gran mayoría de muestras puede producir curvas de fusión interpretables con un tinte de reportero basado en hidrofobicidad o a través de nanoDSF libre de tinte. Si fuentes de fluorescencia estándar no son adecuadas para su proteína, la modificación más simple para el protocolo que podría explorarse es la elección del fluoróforo. Varios tintes alternativos podrían ser adecuados para los experimentos de la TSA. Los ejemplos incluyen N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM), un compuesto que aparece después de reaccionar con un tiol27y 4 – julolidine (dicyanovinyl) (DCVJ), un compuesto que varía su fluorescencia basada en la rigidez de su entorno, aumentando su fluorescencia como una muestra de la proteína despliega28,29 (el tinte de este último a menudo requiere altas concentraciones de la muestra).
Métodos alternativos de análisis de la curva de fusión están disponibles si Tm no es calculado automáticamente por el software del instrumento. Si los datos son homogéneos y solo un paso de desnaturalización es evidente en las curvas de fusión, se puede montar un conjunto de datos truncado un colon sigmoide con la siguiente ecuación de Boltzmann:
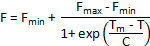
Donde F es la intensidad de fluorescencia a temperatura T, Fmin ymax F son las intensidades de fluorescencia antes y después de la transición de la desnaturalización, respectivamente, Tm la temperatura del punto medio de la transición de desnaturalización y C es la pendiente a Tm. Mientras que este método funciona bien para los procesos de desnaturalización de dos etapas simple, es inadecuado para las curvas de fusión compleja con múltiples transiciones.
Una de las principales ventajas de TSA es su accesibilidad; Experimentos TSA se pueden realizar en cualquier sistema de RT-PCR con filtros de longitudes de onda adecuadas para el colorante de fluorescencia empleado. Esto junto con el bajo costo de consumibles, facilidad de operación y la relativamente baja cantidad de proteína necesaria, hacer TSA una técnica valiosa para una amplia gama de escalas de proyecto, tanto en la industria y la academia.
Así como indicando las condiciones favorables de búfer, las pantallas contienen algunos pozos que pueden dar pistas a la presencia de metales estructurales dentro de una proteína de la muestra. Pozos que pueden ser de interés particular en la pantalla de sal son G6 y G7, que contienen EDTA 5 mM y 5 mM EGTA, respectivamente. Importante desestabilización térmica en estos pozos puede ser indicativo de importantes iones del metal en la proteína que son secuestrados por los chelants. Compuestos dentro de la pantalla del osmolyte también potencialmente pueden dar pistas a la función de una proteína. Muchos de los compuestos en la pantalla pertenecen a las clases de molécula que son sustratos comunes de enzimas. Por ejemplo, la estabilización general de sacáridos (presentes en pozos A11-B10) de lisozima se podía atribuir a su semejanza estructural a establecido los sustratos de la enzima N-acetilglucosamina oligómeros30.
Los protocolos TSA y nanoDSF mencionados también puede adaptarse para el estudio de las interacciones proteína-ligando. Ligandos que se unen específicamente a una proteína pueden aumentar su estabilidad térmica mediante la introducción de nuevas interacciones dentro del complejo. Un cambio positivo dependiente de la dosis de proteína Tm es un signo prometedor de una interacción exitosa proteína-ligando. La velocidad, rendimiento y bajo costo de detección compuestas bibliotecas con TSA ha hecho un método muy popular en el descubrimiento de la droga de la temprano-etapa.
Optimización de las condiciones de buffer de las proteínas Diana y sus complejos ligando puede ser esencial para el éxito de un proyecto, como lo demuestran los muchos ejemplos de literatura31,32,33,34. Con un típico ensayo tomando bajo 2 horas incluyendo tiempo de instalación, TSAs y nanoDSF juntada con las pantallas de estabilidad representan una técnica rápida, de bajo costo para optimizaciones de búfer.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Este proyecto ha recibido financiación del Consejo Europeo de investigación (ERC) programa de investigación e innovación a los horizonte 2020 de la Unión Europea (beca Convenio n ° 685778). Este trabajo fue apoyado por la biotecnología y el Consejo de investigación de ciencias biológicas (BBSRC, grant números M011186/BB/1, J014516/BB/1). DB gracias el BBSRC Doctoral formación Asociación Newcastle-Liverpool-Durham para una beca y Departamento de biociencias de la Universidad de Durham para contribuir a la financiación de este trabajo. Agradecemos a Ian Edwards por su ayuda y el Departamento de la Durham University Departamento de química espectrometría de masas para su análisis instrumental de la proteína X. Agradecemos a Arnthor Ævarsson por su trabajo con el proyecto de Virus X y gracias también a Claire Hatty y NanoTemper GmbH para financiamiento y asistencia con el sistema de Prometheus NT.48 para este proyecto. Por último, gracias a Frances Gawthrop y Tozer semillas por su apoyo como parte del Premio de iCASE BBSRC.
Materials
| Lysozyme | Melford Laboratories | L38100 | Crystallised and lyophilised chicken egg white lysozyme. |
| The Durham pH Screen | Molecular Dimensions | MD1-101 | 96-well protein stability screen. See above for contents. |
| The Durham Salt Screen | Molecular Dimensions | MD1-102 | 96-well protein stability screen. See above for contents. |
| The Durham Osmolyte Screen | Various | #N/A | 96-well protein stability screen, not commercially available at the time of publication. See above for contents. |
| SYPRO Orange | Invitrogen | S6651 | Widely used fluorescent dye for protein staining in gels and DSF. |
| 96-well PCR Plate | Starlab | 1403-7700 | Semi-skirted clear plastic for use with AB 7500 Fast RT-PCR System. |
| 7500 Fast Real-time PCR System | Applied Biosystems | 4362143 | 96-well format RT-PCR system. Alternative systems can be used. Analysis of data performed using free, open-access software NAMI. AB software tailored to DSF experiments using the 7500 Fast available at additional cost. |
| Prometheus NT.48 | NanoTemper Technologies | #N/A | Label-free DSF system with up to 48-sample capacity. Can calculate unfolding temperatures (Tm and Tonset), critical denaturant concentrations (Cm), free folding energy (ΔG and ΔΔG), and aggregation results (Tagg) using built-in software. |
| Prometheus NT.48 Series nanoDSF Grade Standard Capillaries | NanoTemper Technologies | PR-C002 | Prometheus NT.48 Series nanoDSF Grade Standard Capillaries. "High sensitivity" variants are available at a higher cost for use with low-concentration samples (<200 µg ml-1). |
References
- Kapust, R. B., Waugh, D. S. Controlled intracellular processing of fusion proteins by TEV protease. Protein Expression and Purification. 19 (2), 312-318 (2000).
- Cordingley, M. G., Register, R. B., Callahan, P. L., Garsky, V. M., Colonno, R. J. Cleavage of small peptides in vitro by human rhinovirus 14 3C protease expressed in Escherichia coli. Journal of Virology. 63 (12), 5037-5045 (1989).
- Hjorleifsdottir, S., Aevarsson, A., Hreggvidsson, G. O., Fridjonsson, O. H., Kristjansson, J. K. Isolation, growth and genome of the Rhodothermus RM378 thermophilic bacteriophage. Extremophiles. 18 (2), 261-270 (2014).
- Alva, V., Nam, S. Z., Söding, J., Lupas, A. N. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Research. 44, 410-415 (2016).
- McPherson, A. Protein Crystallization. Methods in molecular biology. 1607, 17-50 (2017).
- Ericsson, U. B., Hallberg, B. M., DeTitta, G. T., Dekker, N., Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Analytical Biochemistry. 357 (2), 289-298 (2006).
- Reinhard, L., Mayerhofer, H., Geerlof, A., Mueller-Dieckmann, J., Weiss, M. S. IUCr Optimization of protein buffer cocktails using Thermofluor. Acta Crystallographica Section F Structural Biology and Crystallization Communications. 69 (2), 209-214 (2013).
- Boivin, S., Kozak, S., Meijers, R. Optimization of protein purification and characterization using Thermofluor screens. Protein Expression and Purification. 91 (2), 192-206 (2013).
- Kozak, S., Lercher, L., Karanth, M. N., Meijers, R., Carlomagno, T., Boivin, S. Optimization of protein samples for NMR using thermal shift assays. Journal of Biomolecular NMR. 64 (4), 281-289 (2016).
- Semisotnov, G. V., Rodionova, N. A., Razgulyaev, O. I., Uversky, V. N., Gripas’, A. F., Gilmanshin, R. I. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 31 (1), 119-128 (1991).
- Grøftehauge, M. K., Hajizadeh, N. R., Swann, M. J., Pohl, E. Protein-ligand interactions investigated by thermal shift assays (TSA) and dual polarization interferometry (DPI). Acta Crystallographica Section D: Biological Crystallography. 71, 36-44 (2015).
- Steinberg, T. H., Jones, L. J., Haugland, R. P., Singer, V. L. SYPRO Orange and SYPRO Red Protein Gel Stains: One-Step Fluorescent Staining of Denaturing Gels for Detection of Nanogram Levels of Protein. Analytical biochemistry. 239, 223-237 (1996).
- Burstein, E. A., Vedenkina, N. S., Ivkova, M. N. Fluorescence and the location of tryptophan residues in protein molecules. Photochemistry and Photobiology. 18 (4), 263-279 (1973).
- Haffke, M., Rummel, G., Boivineau, J., Münch, A., Jaakola, V. -. P. nanoDSF: label-free thermal unfolding assay of G-protein-coupled receptors for compound screening and buffer composition optimization. Application Note NT-PR-008. , (2016).
- Chari, A., et al. ProteoPlex: stability optimization of macromolecular complexes by sparsematrix screening of chemical space. Nature Methods. 12 (9), 859-865 (2015).
- Laskowski, R. A. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Research. 29 (1), 221-222 (2001).
- Good, N. E., Winget, G. D., Winter, W., Connolly, T. N., Izawa, S., Singh, R. M. Hydrogen ion buffers for biological research. Biochemistry. 5 (2), 467-477 (1966).
- Good, N. E., Izawa, S. Hydrogen ion buffers. Methods in enzymology. 24, 53-68 (1972).
- Ferguson, W. J., et al. Hydrogen ion buffers for biological research. Analytical biochemistry. 104 (2), 300-310 (1980).
- Welch, W. J., Brown, C. R. Influence of molecular and chemical chaperones on protein folding. Cell stress & chaperones. 1 (2), 109-115 (1996).
- Diamant, S., Eliahu, N., Rosenthal, D., Goloubinoff, P. Chemical chaperones regulate molecular chaperones in vitro and in cells under combined salt and heat stresses. The Journal of biological chemistry. 276 (43), 39586-39591 (2001).
- Yancey, P. H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. Journal of Experimental Biology. 208 (15), 2819-2830 (2005).
- Garman, E. F., Owen, R. L. Cryocooling and radiation damage in macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography. 62 (1), 32-47 (2006).
- de Marco, A., Vigh, L., Diamant, S., Goloubinoff, P. Native folding of aggregation-prone recombinant proteins in Escherichia coli by osmolytes, plasmid- or benzyl alcohol- overexpressed molecular chaperones. Cell Stress & Chaperones. 10 (4), 329 (2005).
- Berman, H. M., et al. The protein data bank. Nucleic acids research. 28 (1), 235-242 (2000).
- Bougouffa, S., Radovanovic, A., Essack, M., Bajic, V. B. DEOP: A database on osmoprotectants and associated pathways. Database. 2014, 1-13 (2014).
- Alexandrov, A. I., Mileni, M., Chien, E. Y. T., Hanson, M. A., Stevens, R. C. Microscale Fluorescent Thermal Stability Assay for Membrane Proteins. Structure. 16 (3), 351-359 (2008).
- Kung, C. E., Reed, J. K. Fluorescent molecular rotors: a new class of probes for tubulin structure and assembly. Biochemistry. 28 (16), 6678 (1989).
- Iio, T., Itakura, M., Takahashi, S., Sawada, S. 9-(Dicyanovinyl)julolidine binding to bovine brain calmodulin. Journal of biochemistry. 109 (4), 499-502 (1991).
- Veros, C. T., Oldham, N. J. Quantitative determination of lysozyme-ligand binding in the solution and gas phases by electrospray ionisation mass spectrometry. Rapid Communications in Mass Spectrometry. 21 (21), 3505-3510 (2007).
- Kean, J., Cleverley, R. M., O’Ryan, L., Ford, R. C., Prince, S. M., Derrick, J. P. Characterization of a CorA Mg2+ transport channel from Methanococcus jannaschii using a Thermofluor-based stability assay. Molecular membrane biology. 25 (8), 653-661 (2008).
- Geders, T. W., Gustafson, K., Finzel, B. C. Use of differential scanning fluorimetry to optimize the purification and crystallization of PLP-dependent enzymes. Acta Crystallographica Section F. 68 (5), 596-600 (2012).
- Morgan, H. P., Zhong, W., McNae, I. W., Michels, P. A., Fothergill-Gilmore, L. A., Walkinshaw, M. D. Structures of pyruvate kinases display evolutionarily divergent allosteric strategies. Royal Society open science. 1 (140120), (2014).
- Moretti, A., Li, J., Donini, S., Sobol, R. W., Rizzi, M., Garavaglia, S. Crystal structure of human aldehyde dehydrogenase 1A3 complexed with NAD+ and retinoic acid. Scientific reports. 6 (35710), (2016).
