The aim of this protocol was to describe the isolation of primary rat VICs and culture them for in vitro calcification experiments. By employing the method described above, rat VICs can be successfully isolated and expanded for the study of the mechanisms responsible for CAVD.
Rat Primary VICs Co-localize with Established VIC Markers:
The VIC phenotype of isolated cells was confirmed through immunofluorescence by probing for the VIC markers: vimentin and α-SMA (red and green, respectively, Figure 1A), and is in agreement with previous reports11,12. The representative negative controls using non-conjugated mouse and rabbit IgG are shown in Figure 1B. Additionally, the expression of the VIC-growth regulator TGFβ-1 and TGFβ-2 was confirmed using PCR analysis (Figure 1C). In order to confirm that the isolated rat primary VICs were free from endothelial contamination, Western blot analysis was performed to verify that rat VICs were negative for the endothelial cell marker, CD31, using canine mitral VECs as a positive control (Figure 1D).
Ca and Pi Induce Rat VIC Calcification:
Elevated systemic Ca and/or Pi concentrations typically drive the calcification of VICs in vitro. To appreciate the calcification potential of the isolated rat VICs, the cells were exposed to elevated levels of Ca and Pi, which mimic pathological hypercalcemia and hyperphosphatemia conditions in ESRD patients. Treatment of VICs with 2.7 mM Ca/2.5 mM Pi induced calcification, as determined by Alizarin Red S staining for Ca deposition (Figure 2A) and colorimetric determination of Ca levels following HCl leaching (81 fold; p < 0.05 using Student's t-Test; Figure 2B).
Gene Expression Changes Associated with VIC Calcification:
The calcification of vascular cells in vitro is associated with a distinct molecular profile. In the present study, the treatment of VICs with 2.7 mM Ca/2.5 mM Pi induced a significant increase in the mRNA expression of the osteogenic markers: Msh homeobox 2, Msx2 (2.04 fold change; p < 0.01; Figure 3A), alkaline phosphatase, Alpl (1.49 fold change; p < 0.001; Figure 3B), and phosphoethanolamine/phosphocholine phosphatase, Phospho1 (4.7 fold change; p < 0.01 using one-way ANOVA; Figure 3C). However, the expression of the osteogenic marker, Runx2, and calcification inhibitor ectonucleotide pyrophasphatase, Enpp1, remained unchanged (Figure 3D–E).
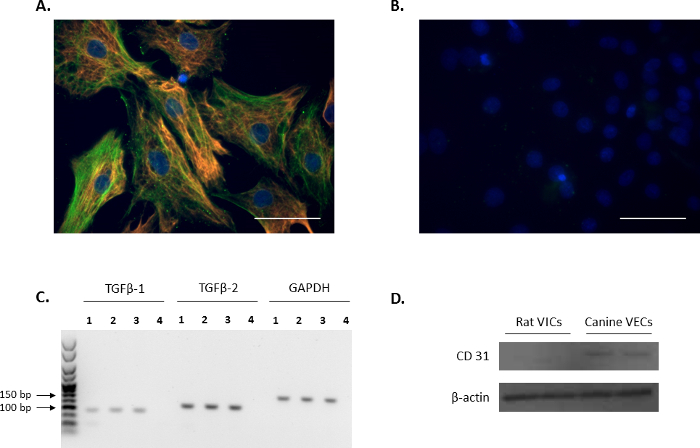
Figure 1. Expression of VIC markers. (A) Immunofluorescence showing double staining and colocalization of alpha-smooth muscle actin (α-SMA; green) and vimentin in valve interstitial cells (VICs). (B) Representative image of negative controls using mouse and rabbit IgG. Nuclei are stained in blue using 4',6-diamidino-2-phenylindole (DAPI). Scale bar = 50 µm. (C) The presence of transforming growth factor beta 1 (TGFβ-1) and TGFβ-2 in VICs (Lanes 1-3) as shown by PCR analysis. Lane 4 is the water control. Reference gene used was Gapdh. (D) Western blot analysis showing the abundant expression of CD31 in canine mitral valve endothelial cells (VECs) in comparison to no expression in VICs. Please click here to view a larger version of this figure.
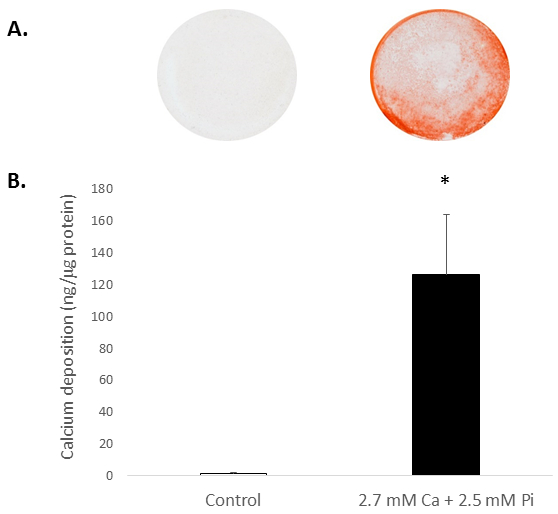
Figure 2. In vitro calcification of rat VICs. Ca deposition in VICs treated with 2.7 mM Ca/2.5 mM Pi as determined by: (A) Photograph showing Alizarin Red S staining of whole cell monolayers in the well, and (B) colorimetric determination of Ca levels following HCl leaching. Student's t-test was performed to analyze the significance between the two data groups. Results are presented as mean ± S.E.M. *p < 0.5 compared with control; (n = 4).
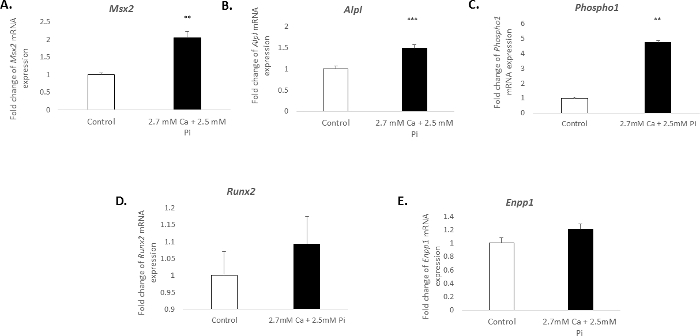
Figure 3. Gene expression changes associated with VIC calcification. Fold change in the mRNA expression of osteogenic markers (A) Msx2, (B) Alpl, (C) Phospho1, (D) Runx2, and (E) Enpp1 in VICs treated with 2.7 mM Ca/2.5 mM Pi for 48 h. mRNA expression is shown as a fold change compared to the endogenous reference gene Gadph. One-way ANOVA using general linear model incorporating pair-wise comparisons was performed to analyze the significance between multiple groups. Results are presented as mean ± S.E.M. **p < 0.01; ***p < 0.001 compared to control; (n = 6). Please click here to view a larger version of this figure.
| Number of valve leaflets | Culture plate/flask |
| 9 to 15 | 1 well in 12-well plate |
| 15 to 30 | 1 well in 6-well plate |
| 30+ | T25 |
Table 1. General guidelines for the number of leaflets required for initial seeding.
