Immunofluorescence Staining Using IBA1 and TMEM119 for Microglial Density, Morphology and Peripheral Myeloid Cell Infiltration Analysis in Mouse Brain
Summary
This protocol describes a step-by-step workflow for immunofluorescent costaining of IBA1 and TMEM119, in addition to analysis of microglial density, distribution, and morphology, as well as peripheral myeloid cell infiltration in mouse brain tissue.
Abstract
This is a protocol for the dual visualization of microglia and infiltrating macrophages in mouse brain tissue. TMEM119 (which labels microglia selectively), when combined with IBA1 (which provides an exceptional visualization of their morphology), allows investigation of changes in density, distribution, and morphology. Quantifying these parameters is important in providing insights into the roles exerted by microglia, the resident macrophages of the brain. Under normal physiological conditions, microglia are regularly distributed in a mosaic-like pattern and present a small soma with ramified processes. Nevertheless, as a response to environmental factors (i.e., trauma, infection, disease, or injury), microglial density, distribution, and morphology are altered in various manners, depending on the insult. Additionally, the described double-staining method allows visualization of infiltrating macrophages in the brain based on their expression of IBA1 and without colocalization with TMEM119. This approach thus allows discrimination between microglia and infiltrating macrophages, which is required to provide functional insights into their distinct involvement in brain homeostasis across various contexts of health and disease. This protocol integrates the latest findings in neuroimmunology that pertain to the identification of selective markers. It also serves as a useful tool for both experienced neuroimmunologists and researchers seeking to integrate neuroimmunology into projects.
Introduction
Whether acute or chronic, neuroinflammation is tightly influenced by microglia, the resident macrophages of the brain. Visualizing microglia through immunostaining is valuable for the study of neuroinflammation with the use of light microscopy, a highly accessible technique. In homeostatic conditions, microglia are typically distributed in a nonoverlapping, mosaic-like pattern. They exhibit small somas that extend ramified processes1, which sometimes contact one another2. Microglial ramified processes dynamically survey the brain parenchyma, interacting with neurons, other glial cells, and blood vessels during normal physiological conditions3. Microglia are equipped with an arsenal of receptors that allow them to perform immunological tasks and respond to changes in the brain milieu, to cell death, or to tissue damage. In addition, they exert key physiological functions, notably in synaptic formation, maintenance, and elimination4,5.
Among the available markers used to study microglia, ionized calcium binding adaptor molecule 1 (IBA1) is one of the most widely used. IBA1 is a calcium binding protein that provides exceptional visualization of microglial morphology, including fine distal processes, as confirmed by electron microscopy6. This tool has been instrumental in characterizing microglial transformation, formerly called "activation", in a vast array of animal disease models7,8,9. In the presence of neuroinflammation, the microglial response includes: microgliosis that is defined as an increase in cellular density, changes in distribution that sometimes result in clustering, enlargement of the cell body, as well as thickening and shortening of processes associated with more ameboid shapes10,11,12,13.
Immunostaining is limited by the availability of antibodies directed against specific markers. Importantly, IBA1 is expressed by microglia but also by peripheral macrophages that infiltrate the brain14. While observation of IBA1-positive cells inside the brain has become a marker of microglia in this research field, peripheral macrophage infiltration has been reported under various conditions, even marginally in the healthy brain15,16,17,18. Consequently, the use of IBA1 alone does not allow selective visualization of microglia. In addition, macrophages adopt molecular and morphological features of resident microglia once they have infiltrated the brain, thus hindering differentiation19. This represents a challenge when investigating the function of both microglia and infiltrating macrophages.
While microglia and peripheral macrophages have distinct origins (e.g., from the embryonic yolk sac and bone marrow, respectively20,21), there is an increasing number of findings indicating that the two cell populations exert different roles in the brain19. It is thus crucial to use methods that discriminate between these two populations without invasive manipulations (i.e., bone marrow chimeras or parabiosis) that can modulate their density, distribution, morphology, and function. TMEM119 has emerged as a microglia-specific marker across health and disease conditions22. When combined with IBA1, this marker becomes useful for differentiating these cells from infiltrating macrophages, which are TMEM119-negative and IBA1-positive. While it is developmentally regulated, TMEM119 is expressed as early as postnatal days 3 (P3) and 6 (P6), steadily increasing until reaching adult levels between P10 and P1422. IBA1 is expressed as early as embryonic day 10.5 (E10.5)23. The proposed double labeling protocol is thus useful to study these two populations throughout postnatal life.
This protocol provides a step-by-step immunostaining procedure that allows discrimination between microglia and peripheral macrophages. It also explains how to conduct a quantitative analysis of microglial density, distribution, and morphology, as well as analysis of peripheral macrophage infiltration. While the investigation of microglia and peripheral macrophages is useful on its own, this protocol further allows localization of neuroinflammatory foyers; thus, it also serves as a platform to identify specific regions to investigate, with the use of complementary (yet, more time- and resource-consuming) techniques.
Protocol
All experimental procedures were performed in agreement with the guidelines of the Institutional Animal Ethics committees, in conformity with the Canadian Council on Animal Care and the Animal Care Committee of Université Laval.
1. Immunostaining
- Select three mouse brain sections containing the region of interest (ROI) (i.e., the hippocampus) with the help of a brain atlas. Place the sections in a plastic multi-well plate and cover them with 350 µL of phosphate-buffered saline (PBS) (Table 1).
NOTE: For optimal results, the brains should be perfused with 4% paraformaldehyde and cut to a thickness of 50 µm with a vibratome. For a 24 multi-well plate, each well can hold up to six sections. The recommended volume of solution for each well is 350 µL (for up to three sections) and 500 µL for wells containing six sections. For a higher number of sections, it is recommended to use a 12 multi-well plate. Make sure that the selected volume of solution for each well completely covers the tissue and allows the sections to float. The recommended volumes apply for every solution used in the rest of the protocol. - Wash the samples by covering them with 350 µL of PBS and let them rest by placing the multi-well plate on top of a multipurpose shaker at room temperature (RT). Remove the PBS after 5 min and replace it 5x with fresh PBS.
NOTE: To remove the solutions, a transfer pipette is recommended. When pouring in any solution, make sure to place the tip of pipette against the well wall to protect tissue integrity. Also make sure to use a new pipette for each new solution. - Remove PBS and add 350 µL of 10 mM sodium citrate buffer with pH = 6.0 (Table 1).
- Seal the multi-well plate with paraffin film and let it float on a previously preheated water bath for 40 min at 70 °C.
- Let the multi-well plate cool down for approximately 15 min.
- Remove the sodium citrate buffer and wash the sections in PBS as done in step 1.2.
- Remove PBS and add 350 µL of freshly made 0.1% NaBH4 (Table 1) and let incubate for 30 min at RT.
- Remove the solution of 0.1% NaBH4 and wash the sections in PBS as done in step 1.2.
- Remove PBS and add blocking buffer (Table 1) for 1 h at RT on top of a multipurpose shaker.
NOTE: Make sure to prepare doubled volumes of blocking buffer, as the same solution will be used in the next step. - Remove the blocking buffer and replace by blocking buffer containing the mixture of primary antibodies (1:150 mouse IBA1 + 1:300 TMEM119). Seal the plate with paraffin film and let it incubate overnight at 4 °C.
- The next day, warm samples at RT for approximately 15 min.
- Wash the sections 5x for 5 min each in PBS with triton (PBST) (Table 1).
- Remove PBST and add blocking buffer containing the mixture of secondary antibodies (1:300 donkey anti-mouse Alexa 488 for IBA1; 1:300 goat anti-rabbit Alexa 568 for TMEM119) for 1.5 h at RT. Starting from this point onward, protect the samples from light.
- Remove blocking buffer and wash the sections 5x as done in step 1.2, except this time with PBST.
- Remove the PBST and add 4′,6-diamidino-2-phenylindole (DAPI) [1:20000] for 5 min at RT.
- Remove DAPI and wash the sections 3x for 5 min each in phosphate buffer (PB).
- Mount the sections on a microscope slide. Let them dry while protected from light.
- When dried, add some drops of mounting fluorescence medium and cover with a coverslip, avoiding bubble formation.
NOTE: Store the slides while protected from light, inside a histological slide box, at 4 °C. The samples can be preserved for several months.
2. Imaging for density and distribution analysis
- With the help of a widefield epifluorescence microscope, use a low magnification and the DAPI channel to locate the ROI (i.e., the CA1 region of the hippocampus).
- Acquire images at 20x, using a numerical aperture (NA) of 0.5, with the DAPI, 488, and 568 channels and filters, at a resolution of 0.3 µm/pixel. Capture a mosaic picture covering the ROI. Alternatively, take individual pictures that will be stitched into a larger image.
NOTE: A mosaic image is a super image constituted by smaller images. Mosaic images are usually used to overcome the limited area of the field-of-view of high magnifications. Some software includes a mosaic function; nevertheless, images can also be manually stitched together with other photo editing software by stitching the individual images into one. Remember to add the scale information to the file. For this type of analysis, it is recommended to have at least 300 microglial cells imaged per ROI/animal (corresponding to approximately 10−15 pictures for the hippocampus, for example), with a minimum of five animals per experimental condition. Figure 1A−C shows the images of colabelled microglia. - Save the image as a TIFF file.
3. Imaging for morphology analysis
- Using a confocal or structured illumination microscope, use the DAPI channel to locate the ROI at low magnification.
- Using a 40x objective (i.e., NA 1.4 oil), locate an IBA1+/TMEM119+ cell inside the ROI. While live imaging, move in the Z-axis. As soon as the signal of the randomly selected microglia disappears, set this Z-level as the beginning of the Z-stack. Move along the Z-axis in the opposite direction until the signal of the microglia disappears and set that point as the end of the Z-stack.
NOTE: Figure 2A−C shows images of IBA1+/TMEM119+ microglia. - Create a Z-stack in all three channels (DAPI, 488, 568) using a 0.33 µm Z-interval and pixel size of 0.15 µm/pixel. Add the scale information to the file.
NOTE: The recommended Z-interval depends on the resolving power of the objective (e.g., for a 40x objective such as NA 1.4 oil, it is 0.33 µm). For morphology analysis, it is recommended to have at least 20 cells per animal with a minimum of five animals per experimental condition. - Save the file as a TIFF file.
4. Density and distribution analysis
- Open FIJI/ImageJ with the nearest neighbor distance (NND) plugin installed. Open the 20x image.
NOTE: Use a search engine with the keyword “Nearest Neighbor Distances Calculation with ImageJ” to find the installation instructions. The plugin Author is Yuxiong Mao. - To set the scale manually based on a scale imprinted on the image, select the straight line tool (Figure 3E), place the cursor on the edge of the scale, and, while pressing the shift key, draw a line as close as possible to the scale on the image (Figure 3I), select Analyze | Set scale, then enter the correct information (Figure 3J).
NOTE: The scale can sometimes be contained in the metadata of the file and set automatically. - Select Image | Color | Make composite to create a composite image of all channels.
NOTE: During image acquisition, FIJI/ImageJ will automatically create a composite in the RGB format. - On the menu bar, select Analyze | Set measurements. Check Area, Centroid, and Perimeter. On the tab Redirect to, click and select the opened file (Figure 3K).
- Go to Image | Color | Channel tool to open the channel tool.
NOTE: This menu will allow a specific color to be disabled. The DAPI channel can be useful to identify ROI and to confirm cells. It can be deactivated to make counting easier. - Draw a rough perimeter of the ROI with the freehand selection tool (Figure 3D).
- Enable the selection brush tool by double-clicking the oval tool on the tool bar and make sure that the Enable selection brush box is checked (Figure 3G). This tool will be used to delineate the ROI more precisely. Select an appropriate brush size between 200−400.
- Using the selection brush, adjust the perimeter to best fit the ROI. Press T on the keyboard to add to the ROI manager (Figure 3L).
- Select Analyze | Measure or press the M key, and a results window will pop up. Copy and paste the results on a datasheet, then save the information regarding the area (i.e., the area of the ROI; Figure 3R).
- After copying the area of the ROI, erase the information from the results window by clicking on it and pressing the Backspace key.
- Go to the ROI manager window (Figure 3L), right-click the ROI trace, change the name to match the image’s name, then save.
- Double-click the brush tool at the tool bar. Select the black color and a brush size of 10. Make sure that the option Paint of overlay is unchecked (Figure 3H).
- In the TMEM119 channel, carefully place a black dot on the center the soma for each TMEM119+ microglia. Place a white dot on the center of the cells that are not positive for TMEM119 (to mark infiltrating macrophages). Repeat the same procedure for all cells contained in the ROI.
NOTE: It is important that all dots (black and white) are located in the same channel. The identity of the channel can be verified (red, blue, or green) by looking at the color of the image window labels. - Select Image | Color | Split channel. A window for each channel will appear. Then, identify the channel that has the dot annotations and close the other two windows.
- Redirect the new split channel image. Go to Analyze | Set measurements. On the tab Redirect to, click and select the split channel image (Figure 3K).
- Select Image | Tipo | 8-bit. Go to Image | Adjust and select Threshold (Figure 3O). To adjust the threshold, slide the button of the second bar, all the way to the left (threshold value = 0) in both bars.
NOTE: This will leave only the black dots on the image, appearing white. - Select the ROI in the ROI manager window. Select Analyze | Analyze particle (Figure 3N). On Size (inchˆ2): write 1−20. Keep the pixel unit unchecked, check Display, summarize and add to manager, and press Ok. The summary window will pop up and give the number of points (Figure 3P). Copy and paste the information to the datasheet.
- Select Plugins | NND. The NND window will pop up (Figure 3Q). Copy/paste all the information to the datasheet. Each number represents the distance each microglia has to the nearest neighboring microglia.
- Go back to the threshold window and slide the first bar all the way to the right (threshold value = 255 in both bars), which will leave all the white dots visible, appearing white (Figure 3M).
- Select Analyze | Analyze particle. The summary window that provides the number of points will pop up (Figure 3P). Copy and paste the information to the datasheet.
- Go to the ROI manager select all the points, right-click, and save with the image’s name. This will allow saving of all the points in a zip file (Figure 3L). Select File | Save as, and save the file with a name that allows identification of the analyzed image.
- Obtain the density of microglia (for each image) by dividing the number of IBA1+/TMEM119+ double-positive cells by the area of the ROI.
NOTE: The values for each picture can be averaged for each animal. The data can then be presented as mean ± standard error of the mean (SEM) of all the animals. - Determine the NND by obtaining an average per picture of the NND values of all TMEM119+ cells.
NOTE: The data can then be presented as mean ± SEM of all the animals. - Calculate the spacing index using the formula: NND2 x density.
NOTE: The data can then be presented as mean ± SEM of all the animals. The units for this measurement will be arbitrary units. - Quantify microglial clusters by identifying cells that have an NND under 12 µm.
NOTE: Here, 12 µm is selected, as it is the approximate distance between two directly juxtaposing microglial cells touching each another with arborizations. If there are more than three microglia that meet this condition, return to the image and verify whether these cells are part of one or multiple clusters. - After confirming the number of clusters, write the number of clusters in the datasheet.
NOTE: The number of clusters can be divided by the ROI area to obtain the density of cells/mm2 for each animal. The data can then be presented as mean ± SEM of all the animals. - To determine the percentage of peripheral myeloid cell infiltration, calculate the % of IBA1+/TMEM119- cells over the total number of myeloid cells (TMEM119+/IBA1+ + TMEM119-/IBA1+) for each animal.
NOTE: The data can then be presented as mean ± SEM of all the animals.
5. Morphology analysis
- Open FIJI/ImageJ.
- Open the 40x image using Image J or FIJI. Select A popup window will appear asking if the images should be opened in a stack. Click OK. Next, select Image | Stacks | Z project to open the Z-Projection window. Include all slices, from the first through last slice. Ensure that the Max Intensity is selected under Projection Type, and click OK
- Click on the new window with the Z project. Select Image | Colors | Split channels. Conduct the traces on the images of the IBA1 channel.
NOTE: The other channels (TMEM119 and DAPI) can be kept open and consulted as needed during the microglial morphology analysis. - On the menu bar, select Analyze | Set measurements. Check the Area, Centroid, and Perimeter. On the tab Redirect to, select the opened file (Figure 3K).
- Set the scale as described in steps 4.2.
- To measure the soma size in the IBA1 channel, draw a rough perimeter of the soma with the freehand selection tool (Figure 3D).
- Enable the selection brush tool by double-clicking the oval tool on the tool bar, followed by checking Enable the selection brush box (Figure 3G). Select a selection brush size between 10−20 (Figure 3B).
- Using the selection brush, adjust the trace to best fit the soma. Zooming in will enable precision during this step (Figure 2I).
- Press the T key to add the soma trace to the ROI manager (Figure 3L).
- Select Analyze | Measure or press the M key. A results window will pop up. Copy and paste the results on a datasheet (Figure 3R).
- To save the information regarding the soma area, go to the ROI manager window, right-click on the ROI, change the name to match the image’s name, specify that the trace is for soma, then save the file.
- To measure arborization area in the IBA1 channel, click on a microglial process extremity with the polygon selection tool, which will start the polygon shape (Figure 3C).
- Following the tips of the microglial processes, go around the microglia by clicking at the tips of each process extremity to form a polygon that best represents the area covered by the microglial arborizations (Figure 2D−H).
NOTE: Make sure that the polygon connects all the microglial process extremities. The lines forming the polygon should never intersect. When clicking around a microglial process tip, be careful to avoid cutting off any part of the process. It is sometimes useful to add extra points to go around a process. The number of points forming the polygon is not directly linked to the number of distal processes and thus is not relevant for the study. - To close the polygon, click on the starting point of the polygon.
- Press the T key to add the trace to the ROI manager (Figure 3L). Select Analyze | Measure or press the M key. A results window will pop up. Copy and paste the results on a datasheet (Figure 3R).
- To save the information regarding the arborization area, go to the ROI manager window, right-click the ROI, change the name to match the image’s name, specify for arborization, then save the file.
- Determine the soma area by averaging all soma areas for each animal.
NOTE: The data can be presented as mean ± SEM of all the animals. - Determine arborization area by averaging all the arborization areas for each animal.
NOTE: The data can be presented as mean ± SEM of all the animals. - Calculate the morphology index by using the formula soma area/arborization area for each microglial cell and average per animal.
NOTE: The data can be presented as mean ± SEM of all the animals.
Representative Results
Figure 1 shows the co-labeling of microglia using IBA1 and TMEM119 in a coronal section of the dorsal hippocampus imaged at 20x by fluorescence microscopy. A successful staining reveals microglial cell bodies and their fine processes (Figure 1A−C). This staining allows determination of microglial density and distribution and identification of microglial clusters (Figure 1I) and infiltrating macrophages (Figure 1F).
Figure 2 shows IBA1+/TMEM119+ microglia (Figure 2A−C) in a stepwise example of the microglial arborization tracing procedure (Figure 2D−H), as well as an example of cell body tracing (Figure 2I), both imaged at 40x by confocal microscopy.
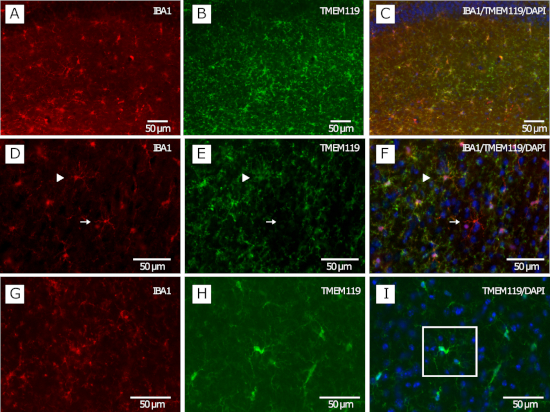
Figure 1: IBA1 and TMEM119 double staining of mouse brain tissue for density, distribution, clustering, and peripheral myeloid cell infiltration analysis. (A−C) Typical microglial distribution in the hippocampus of a C57BL/6 adult mouse. (D−F) Microglia identified as IBA1+/TMEM119+ and infiltrating macrophage identified as IBA1+/TMEM119 (white arrow) in the amygdala of a male mouse. (G−I) Cluster of two microglia (white square) in the hippocampus of a mouse. Please click here to view a larger version of this figure.
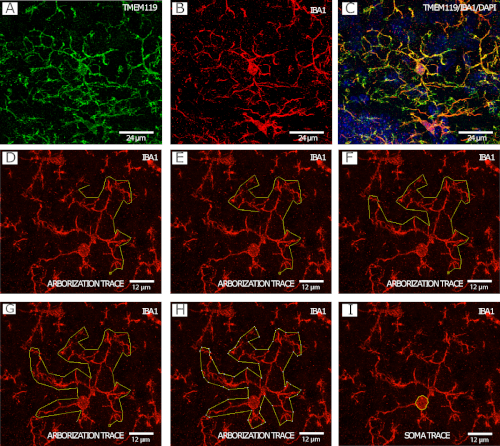
Figure 2: IBA1 and TMEM119 staining for microglial morphology analysis. (A−C) Microglia. (D−I) Step-by-step example of arborization tracing with the IBA1 channel using the polygon tool in FIJI/ImageJ. (J) Example of microglia soma tracing with the IBA1 channel using the freehand selection tool in FIJI/ImageJ. Please click here to view a larger version of this figure.
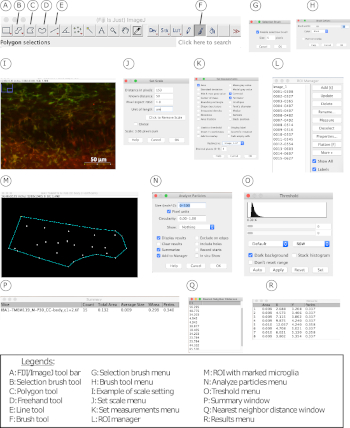
Figure 3: FIJI/ImageJ interface and tools for microglial density, distribution, clustering, morphology, and peripheral myeloid cell infiltration analysis. (A−R) Compilation of all tools, menus, and windows used for the density, cluster, and morphology analyses. Please click here to view a larger version of this figure.
| Solutions | Preparation |
| Blocking buffer | 0.5% gelatin + 5% natural goat serum + 5% natural donkey serum + 0.01% Triton X-100 in TBS [0.05 M] |
| Citrate buffer | 1.92 g of citric acid [10 mM], 500 µL of Tween 20 [0.05% (v/v)], 700 mL of ultrapure water, adjust pH = 6.0 with NaOH [10 N], fill up to 1 L with ultrapure water |
| NaBH4 | [0.01% w/w] Disolve 0.01 g of NaBH4 in 10 mL of ultrapure water, the solution should be well mixed. This solution creates bubble; release pressure by opening the cap after mixing |
| PB | [100 mM] Disolve 23.48 g of Na2HPO4 and 4.8 g of NaH2PO4·H2O in 1 L of ultrapure water, then fill up to 2 L, adjust pH = 7.4 |
| PBS | [50 mM] Disolve 5.87 g of Na2HPO4, 1.2 g of NaH2PO4·H2O, 9 g of NaCl in 500 mL of ultrapure water, fill up to 1 L with ultrapure water, adjust pH = 7.4 |
| PBST | PBS + 0.01% Triton X-100 |
| TBS | Dilute Tris HCl [0.5 M] with ultrapure water 1:10 [0.05 M], take 1 L of Tris HCl [0.05 M] and add 8.75 g of NaCl |
| Tris HCl | [0.5 M] 950 mL of ultrapure water, add 78.8 g of Tris buffer hydrochloride (C4H11NO3Cl) adjust pH = 8 and fill up to 1 L |
Table 1: Solutions used for immunostaining.
Discussion
This protocol can be divided in two critical parts: quality of the staining and analysis. If the staining is not optimal, it will fail to represent microglial cells adequately, thus affecting the density, distribution, and morphology measurements. In addition, the proportion of infiltration peripheral myeloid cells may be underestimated. This is an optimized version of the staining protocol, but there are several factors that may result in suboptimal images. Even though the perfusion of the animal is not included in this protocol, if brain fixation is not well-executed, the quality of the staining will be compromised. Additionally, sufficient perfusion is required to ensure the absence of macrophages inside of blood vessels that may interfere with the study.
With regards to immunostaining, the most critical details include the quality of buffers, blocking step, proper storage of antibodies, and brain sample handling. The proper preparation of buffers and their storage has a direct influence on quality of the staining. Unless specified, some buffers can be stored for long periods, but the use of any buffer that shows signs of contamination should be avoided. If buffers are prepared days or weeks in advance, the pH of every solution before use should be verified.
Additionally, regarding immunostaining, the presence of background staining remains one of the most common problems. Background staining makes it difficult to analyze microglia, especially their morphology, and hence will bias the results. To prevent background, it is important that the blocking step is done correctly. The storing conditions of the antibodies also have direct effects on their efficacy. It is advised to strictly follow the storage guidelines provided by the company as well as avoid frequent thawing-freezing cycles. Finally, during the whole process, it is critical to pay attention to the physical integrity of the brain sections. It is important to use caution during each manipulation (buffer changes, washes, and mounting), especially if the experimenter is not experienced with this procedure. It is advised to avoid leaving the samples without any liquid solution when changing solutions or buffers, solutions for the subsequent step should be ready to pour into the well beforehand. The multi-well plate should be correctly sealed with paraffin film during the overnight step to avoid evaporation that may lead the samples to dry.
Quantitative analysis of microglial density, distribution, and morphology has several advantages over qualitative reports. To prevent bias, the researcher performing the analysis should be blinded to the experimental condition. Thus, it is suggested to have different people perform the analysis and change the name of the files (while keeping the original and new names in a key sheet). The new names should have no hints of the experimental condition. The entire analysis can be done on these blinded files, and the original image identity is revealed only after the compilation of data and prior to statistical analysis. Although blinding is already practiced by experienced researchers, it remains valuable advice for those performing this type of analysis for the first time.
Controlling for the brain region is done during brain section selection and tracing of the ROI during analysis. Make sure to use sections from the same range of Bregma levels across animals. The same ROI should be used for the density, distribution, and morphology analyses. For density and distribution analyses, it is particularly important to be precise when drawing the ROI in FIJI/Image J. The use of a brain atlas is strongly recommended for both section selection and ROI tracing. The use of DAPI also facilitates the identification of neuroanatomical landmarks. To avoid variance, it is recommended to reject microglia that are only partially located in the ROI, as they may differ among their microenvironment. When marking microglia for density analysis, the DAPI channel can be used as a selection criterion. By only counting microglia that contain DAPI-stained nuclei, all considered microglia are in the same plane, reducing the personal bias during selection.
Since measurements for the NND, spacing index, and cluster analysis are based on the locations of dots marking individual cells, and since the distances are calculated by FIJI/ImageJ, it is important to be consistent when placing these dots. Make sure to strictly place the dots in the center of the cell body, which is determined visually. Additionally, the size of the dots should remain consistent throughout the analysis. This will contribute to a better representation of the spatial distribution of the microglial population. For cluster analysis, 12 µm was selected as a distance threshold based on our previous analyses. Nevertheless, if there are four or more different cells with an NND below 12 µm, all these cells could take part of a single cluster or represent two clusters of two cells. This made it necessary to return to the images and confirm the actual number of clusters.
Unlike density and distribution, in which the ROI is determined by neuroanatomical features using a brain atlas, the selection of microglial cells for morphology analysis is based on the ability to analyze the cell. All the cells that can be analyzed should be selected for analysis in a Z-stack before moving to another Z-stack to prevent selection bias. Reasons for excluding cells include issues with the immunostaining or tissue cutting, processing (e.g., tearing), or mounting (e.g., bubble formation). Ideally, brain sections with such issues should be systematically excluded from imaging and analysis. It is also important to note that the staining for TMEM119 and IBA1 does not show 100% overlap (Figure 2A−C). Because TMEM119 does not allow visualization of process continuity (as well as IBA1), this makes it difficult to assess where one cell ends and where another one starts. Thus, the morphology analysis is done using the IBA1 channel. Additionally, all traces and dots should be saved and visualized for future revision, allowing for increasing transparency and reproducibility of results.
This protocol provides valuable information regarding microglia and infiltrating macrophages. Examples of its applications include detecting signs of neuroinflammation through changes in microglia in different brain regions, studying the anti-inflammatory effects of a compound, and studying factors that interfere with the proper function of microglia. Considering that this protocol allows detection of infiltrating macrophages in the brain and differentiation of these cells from microglia, additional applications include: determination if the recruitment of macrophages occurs in a particular insult or with the use of other techniques (i.e., genetic tools), and confirming and studying the consequences of the absence of peripheral macrophages in the brain during insult. Keep in mind that fluorescence microscopy on its own is not sufficient to confirm infiltration inside the brain parenchyma. When IBA1+/TMEM119- cells are observed near the ventricles or perivascular space, higher spatial resolution techniques such as electron microscopy are required to confirm their localization within the parenchyma. While changes in density, distribution, and morphology are good indicators of microglial and macrophage roles, this approach is most powerful when combined with functional investigations.
Declarações
The authors have nothing to disclose.
Acknowledgements
We are grateful to Nathalie Vernoux for her guidance and assistance with the experiments. We would also like to thank Drs. Emmanuel Planel and Serge Rivest for the use of their fluorescence and confocal microscopes, respectively. This work was partly funded by scholarships from Mexican Council of Science and Technology (CONACYT; to F.G.I), Fondation Famille-Choquette and Centre thématique de recherche en neurosciences (CTRN; to K.P.), Fonds de Recherche du Québec – Santé (to M.B.), and Shastri Indo-Canadian Institute (to K.B.), as well as a Discovery grant from Natural Sciences and Engineering Research Council of Canada (NSERC) to M.E.T. M.E.T. holds a Canada Research Chair (Tier II) of Neuroimmune Plasticity in Health and Therapy.
Materials
| Alexa Fluor 488 donkey anti-mouse | Invitrogen/Thermofisher | A21202 | |
| Alexa Fluor 568 goat anti-rabbit | Invitrogen/Thermofisher | A11011 | |
| Biolite 24 Well multidish | Thermo Fisher | 930186 | |
| Bovine serum albumin | EMD Millipore Corporation | 2930 | |
| Citric acid | Sigma-Aldrich | C0759-500G | |
| DAPI Nuceleic acid stain | Invitrogen/Thermofisher | MP 01306 | |
| Fine Brush | Art store | ||
| Fluoromount-G | Southern Biotech | 0100-01 | |
| Gelatin from coldwater fish skin | Sigma-Aldrich | G7765 | |
| Microscope coverglass | Fisher Scientific | 1254418 | |
| Microslides positively charged | VWR | 48311-703 | |
| Monoclonal mouse Anti-IBA1 | Millipore | MABN92 | |
| Na2H2PO4·H2O | BioShop Canada Inc. | SPM306, SPM400 | |
| Na2HPO4 | BioShop Canada Inc. | SPD307, SPD600 | |
| NaBH4 | Sigma-Aldrich | 480886 | |
| NaCl | Fisher Scientific | S642500 | |
| Normal donkey serum (NDS) | Jackson ImmunoResearch laboratories Inc. | 017-000-121 | |
| Normal goat serum (NGS) | Jackson ImmunoResearch laboratories Inc. | 005-000-121 | |
| Parafilm-M | Parafilm | PM-999 | |
| Rabbit monoclonal Anti-TMEM119 | Abcam | ab209064 | |
| Reciprocal Shaking bath model 25 | Precision Scientific | – | |
| Transfer pipette | |||
| Tris buffer hydrochloride | BioShop Canada Inc. | TRS002/TRS004 | |
| Triton-X-100 | Sigma-Aldrich | T8787 | |
| Tween 20 | Sigma-Aldrich | P7949-100ML |
Referências
- Lawson, L. J., Perry, V. H., Dri, P., Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neurociência. 39 (1), 151-170 (1990).
- Milior, G., et al. Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain, Behavior, and Immunity. 55, 114-125 (2016).
- Nimmerjahn, A., Kirchhoff, F., Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science. 308 (5726), 1314-1318 (2005).
- Hickman, S., Izzy, S., Sen, P., Morsett, L., Khoury, J. E. Microglia in neurodegeneration. Nature Neuroscience. 21 (10), 1359 (2018).
- Tay, T. L., Savage, J. C., Hui, C. W., Bisht, K., Tremblay, M. &. #. 2. 0. 0. ;. Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. The Journal of Physiology. 595 (6), 1929-1945 (2017).
- Tremblay, M. &. #. 2. 0. 0. ;., Lowery, R. L., Majewska, A. K. Microglial Interactions with Synapses Are Modulated by Visual Experience. PLoS Biology. 8 (11), (2010).
- Jakovljevic, M., et al. Induction of NTPDase1/CD39 by Reactive Microglia and Macrophages Is Associated With the Functional State During EAE. Frontiers in Neuroscience. 13, (2019).
- Taylor, A. M. W., et al. Microglia Disrupt Mesolimbic Reward Circuitry in Chronic Pain. The Journal of Neuroscience. 35 (22), 8442-8450 (2015).
- Poliani, P. L., et al. TREM2 sustains microglial expansion during aging and response to demyelination. The Journal of Clinical Investigation. 125 (5), 2161-2170 (2015).
- Lu, S. M., et al. HIV-1 Tat-Induced Microgliosis and Synaptic Damage via Interactions between Peripheral and Central Myeloid Cells. PLoS ONE. 6 (9), e23915 (2011).
- Rodríguez, J. J., et al. Increased densities of resting and activated microglia in the dentate gyrus follow senile plaque formation in the CA1 subfield of the hippocampus in the triple transgenic model of Alzheimer’s disease. Neuroscience Letters. 552, 129-134 (2013).
- Rasmussen, S., et al. Persistent activation of microglia is associated with neuronal dysfunction of callosal projecting pathways and multiple sclerosis-like lesions in relapsing-remitting experimental autoimmune encephalomyelitis. Brain. 130 (11), 2816-2829 (2007).
- Walker, F. R., et al. Dynamic structural remodelling of microglia in health and disease: a review of the models, the signals and the mechanisms. Brain, Behavior, and Immunity. 37, 1-14 (2014).
- Ohsawa, K., Imai, Y., Kanazawa, H., Sasaki, Y., Kohsaka, S. Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. Journal of Cell Science. 113 (17), 3073-3084 (2000).
- Yamasaki, R., et al. Differential roles of microglia and monocytes in the inflamed central nervous system. Journal of Experimental Medicine. 211 (8), 1533-1549 (2014).
- Wohleb, E. S., et al. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology. 37 (9), 1491-1505 (2012).
- Shemer, A., et al. Engrafted parenchymal brain macrophages differ from microglia in transcriptome, chromatin landscape and response to challenge. Nature Communications. 9, (2018).
- Geissmann, F., et al. Development of monocytes, macrophages and dendritic cells. Science (New York, N.Y). 327 (5966), 656-661 (2010).
- Minogue, A. M. Role of infiltrating monocytes/macrophages in acute and chronic neuroinflammation: Effects on cognition, learning and affective behaviour. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 79, 15-18 (2017).
- Ginhoux, F., et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science (New York, N.Y). 330 (6005), 841-845 (2010).
- Kierdorf, K., et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nature Neuroscience. 16 (3), 273-280 (2013).
- Bennett, M. L., et al. New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America. 113 (12), E1738-E1746 (2016).
- Mizutani, M., et al. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. Journal of Immunology. 188 (1), 29-36 (2012).
