NOTE: The overall protocol has been numbered according to folders that will be created and named in step 1.2 (Figure 1 and 2). This protocol represents a standard comparative de novo transcriptome analysis, and every step detailed here may not be necessary for all researchers. This workflow is documented thoroughly on a companion tutorial wiki, which also contains all additional files and links to documents of interest 3rd party developers for each analysis package (Table 1). Links to this material will be included throughout this protocol for easy access to this information. Best practices are notes provided to users as suggestions for the best way to accomplish tasks or for users to consider, and will be communicated through notes in the protocol. A folder of example data input and analytical output is publicly available to users, and is organized as suggested in the protocol (de novo transcriptome assembly and analysis.
1. Set up the Project, Upload Raw Sequencing Reads, and Assess Reads Using FastQC
- Get access to Atmosphere and the Discovery Environment.
- Request a free CyVerse account by navigating to the registration page (e.g. person@institution.edu).
- Fill in the required information and submit.
- Navigate to the main webpage (http://www.cyverse.org/), and select "Sign In" at the top toolbar. Select "Cyverse Login" and sign in using your CyVerse credentials.
- Navigate to the Apps & Services tab, and request access to Atmosphere. Access to the Discovery Environment is automatically granted.
- Set up the project and move data to the Data Store.
- Log into the Discovery Environment (https://de.iplantcollaborative.org/de). Select the “Data” tab to bring up a menu containing all the folders in the Data Store.
- Create a main project folder that will house all of the data associated with the project. Find the toolbar at the top of the data window and select File | New Folder. Do not use spaces or special characters in the folder names or any input/output file names e.g. "!@#()[]{}:;$%^&*." Instead, use underscores or dashes, i.e. "_" or "-" where appropriate.
- Create five folders within the main project folder to organize analyses (Figure 1) Name the folders as follows without commas or quotation marks: "1_Raw_Sequence," "2_High_Quality_Sequence," "3_Assembly," "4_Differential_Expression," "5_Annotated_Assembly." Subfolders will be placed into each of these main project folders (Figure 2).
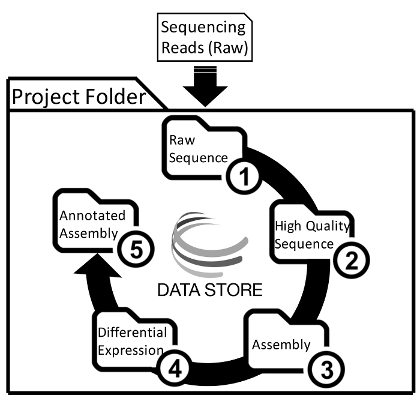
Figure 1: A General Overview of Project Folder Organization and the De Novo Transcriptome Assembly and Analysis Workflow. Users will upload raw sequencing reads into the main project folder on the Data Store, and then place the results from each step into separate folders. Please click here to view a larger version of this figure.
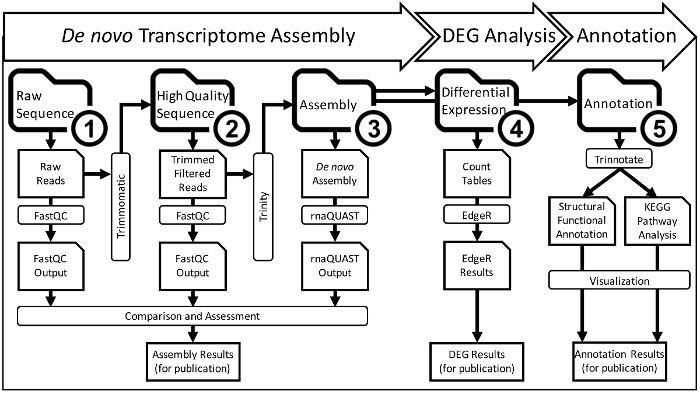
Figure 2: A Detailed Overview of the De Novo Transcriptome Assembly and Analysis Workflow that Occurs within CyVerse Cyberinfrastructure. The entire assembly and analysis workflow will be completed in five steps which each get their own folder (bolded, numbered folder icons). Each of the five numbered workflow step folders has subfolders containing output data from bioinformatic analyses (folder icons). Inputs for analysis come from one subfolder and then move into another folder through the output of an analysis program (rectangle boxes). The final data from the first three steps is compared and prepared for publication. Ultimately, this scheme yields a main project folder that has stepwise analysis for collaborators and/or manuscript reviewers can quickly understand the workflow and repeat it using each file if necessary. Please click here to view a larger version of this figure.
- Upload raw FASTQ sequence files into the folder "1_Raw_Sequence" into a subfolder entitled "A_Raw_Reads" using one of the following three methods.
- Use the Data Store simple upload feature to navigate to the Data window toolbar by clicking on the data button in the main DE desktop, and select Upload | Simple Upload from Desktop. Select the Browse button to navigate to the raw FASTQ sequencing files on the local computer. This method is only suitable for files under 2 GB.
- Select the Upload button at the bottom of the screen to submit the upload. A notification will register in the top right of the DE in the bell icon that the upload has been submitted. Another notification will register when the upload is complete.
- Alternatively, use Cyberduck to transfer larger files (https://wiki.cyverse.org/wiki/x/pYcVAQ). Install Cyberduck and then run as a program on the local computer’s desktop.
- Lastly, download iCommands and install onto the local computer according to instructions (https://wiki.cyverse.org/wiki/display/DS/Using+iCommands).
- Assess uploaded, raw sequencing reads using the FastQC app in the DE.
- Select the "Apps" button on the main DE desktop to open a window containing all of the analysis apps available in the DE.
- Search and open the window for the FastQC tool in the search toolbar at the top of the window. Open the multi-file version if there is more than one FASTQ file. Select File | New Folder to create a folder named "B_FastQC_Raw_Reads" and select this folder as the output folder.
- Load the FASTQ read files into the tool window called "Select input data" and select "Launch Analysis."
- Open the .html or .pdf file to view the results once the analysis is complete. FastQC runs several analyses that test different aspects of the read files (Figure 3).
2. Trim and Quality Filter Raw Reads to Yield High Quality Sequence
Note: Use either the Trimmomatic app or the Sickle app.
- Search for the programmable Trimmomatic app in the DE and open it as before.
- Upload the folder of raw FASTQ read files into the "Settings" section.
- Select whether the sequencing files are single- or paired-end.
- Use the standard control file provided by selecting the Browse button and pasting /iplant/home/shared/Trinity_transdecoder_trinotate_databases into the "Viewing:" box. Select the file named Trimmomaticv0.33_control_file and launch the analysis. The file can be downloaded, the settings edited, and then uploaded into the second project folder to create a custom trimming script.
- Optional: If the FastQC analysis identified adapter sequences, use the ILLUMINACLIP setting to trim Illumina adapters. Select the appropriate adapter file in the folder /iplant/home/shared/Trinity_transdecoder_trinotate_databases as above.
- Quality trimming sequence reads using Sickle.
- Search and open the Sickle app in the DE. Select the trimmed FASTQ reads as input reads, and rename output files. Include quality settings in the options. Typical settings are Quality format: illumina, sanger, solexa; Quality threshold: 20; Minimum length: 50.
- Move all output into the trimmed and filtered folder (2_High_Quality_Sequence).
- Assess the final reads using FastQC and compare to previous FastQC reports. Select the .html file to bring up a webpage of all results. Select the folder of image files (.png) that are provided in the output if that cannot be viewed.
3. De Novo Transcriptome Assembly Using Trinity in Atmosphere
- Open the most current version of the Atmosphere instance by navigating to the wiki page (https://wiki.cyverse.org/wiki/x/dgGtAQ). Select the link for the most recent version of the Trinity and Trinotate image. Alternatively, search “Trinotate” in the Atmosphere image search tool (https://atmo.iplantcollaborative.org/application/images) to bring up all versions of the Trinity and Trinotate images.
- Select the "Log in to launch" button and then name the Atmosphere instance.
- Select an instance size of either "medium3" (CPU: 4, Mem: 32GB) or "large3" (CPU: 8, Mem: 64 GB). Launch the instance, and wait for it to build. In some rare cases CyVerse undergoes maintenance to update platforms. Existing instances are available during these updates, but it may not be possible to create new instances. Visit the CyVerse Status page to see the current state of any platform ( http://status.cyverse.org/ ).
- Open the instance once it is ready by clicking on the name and then selecting "Remote Desktop" on the bottom of the menu on the right. Allow Java and VNC Viewer if asked. Select the "Connect" button in the VNC Viewer window, and then select "Continue."
- Log in to open a separate window that will be the new cloud computing instance.
- Move the trimmed and/or filtered FASTQ read files into the instance using one of the three methods described in steps 1.3.1 – 1.3.4. Use the Internet browser to access the DE and download files just as before on the local computer. Or use iCommands installed on these images to quickly transfer large data sets.
- Running Trinity to assemble high quality reads.
- Set up the analysis folder on the Atmosphere instance. Use the script available in the DE (/iplant/home/shared/Trinity_transdecoder_trinotate_databases) or copy and paste the commands from wiki page (https://wiki.cyverse.org/wiki/x/dgGtAQ). Explanation of all commands can be found on the wiki page.
- Once the analysis folder and the Trinotate databases are established, run the Trinity assembler using the commands from above. There are several output files, but the most important is the final assembly file entitled "Trinity.fasta." Rename this FASTA file to be unique to the organism and treatment of the assembled reads before moving it into the Data Store (folder 3_Assembly) to minimize potential confusion.
NOTE: Output counts tables for differential gene expression analysis into a folder (4_Differential_Expression).
- Assess the assembly using rnaQUAST (Figure 4).
- Move the Trinity output files into the folder "3_Assembly" in the DE and label the folder "A_Trinity_de_novo_assembly." Give each transcriptome that was assembled a subfolder inside the "A_Trinity_de_novo_assembly" folder with unique names including the scientific name of organisms and treatments associated with each transcriptome. Create another subfolder called "B_rnaQUAST_Output" in the "3_Assembly folder."
- Open the app titled "rnaQUAST 1.2.0 (denovo based)" and name the analysis and select "B_rnaQUAST_Output" as the output folder.
- Add the de novo assembly FASTA file(s) to the "Data Input" section. In the "Data Output" section, type a unique name for the de novo assembly. This will create a folder of rnaQUAST output files inside of the folder "B_rnaQUAST_Output."
- Select additional options in the "GenemarkS-T Gene Prediction," "BUSCO," and "Parameters" sections.
- Select prokaryote in the "GenemarkS-T Gene Prediction" section if the organism is not eukaryotic.
- Run BUSCO to select the browse button and copy the path iplant/home/shared/iplantcollaborative/example_data/BUSCO.sample.data into the "Viewing:" box and press enter. Select the most specific BUSCO folder that is available for the organism.
NOTE: BUSCO will assess the assembly for lineage-specific core genes, and output what percentage of core genes is found. There are general folders, e.g. eukaryote, and more specific lineages, e.g. arthropoda.
- Search for "Transcript decoder" and run Transdecoder on the de novo Trinity assembly output FASTA file in the Discovery Environment.
- Move the output .pep file into the de novo assembly (3_Assembly) folder for use in step 5 Annotation.
4. Pairwise Differential Expression Using DESeq2 in the DE
- Open the DESeq2 app in the DE as described previously. Name the analysis and select the output folder as 4_Differential_Expression.
- In the "Inputs" section, select the counts table file from the Trinity assembly run and the column that the contig names can be found in that counts table.
- Input the column headers from the counts data table file to determine which columns are compared. Include the commas between each of the conditions. Do not include the first column header that contains the contig names.
- For replicates, repeat the same name (e.g., Treatment1rep1, Treatment1rep2, Treatment1rep3 would become Treatment1, Treatment1, Treatment1). In the second line, provide the names of the two conditions to be compared (e.g., Treatment1, Treatment2). Match the column header names provided in the first line.
NOTE: These column headers must be alphanumeric and cannot contain any special characters.
5. Annotation Using Trinotate
- Run each part of Trinotate in the Atmosphere cloud computing instance. Note: Bash commands are provided in a txt file to be copied, pasted, and then modified before running on the DE (/iplant/home/shared/Trinity_transdecoder_trinotate_databases) or on the wiki page (https://wiki.cyverse.org/wiki/x/dgGtAQ). If annotating multiple assemblies, annotate each assembly one at a time and then transfer completed annotations files back to folder “5_Annotation” each with a unique folder corresponding with the assembly name.
- Run the bash command for searching Trinity transcripts. Change the number of threads to match how many CPUs are on the instance, i.e. medium has 4 CPUs and large has 8 CPUs. Refer to step 3.1.2 for more details. Change the command Trinity.fasta to match the assembly FASTA file name.
NOTE: BLAST+ searches will require the most time. It may be days before it completes. The cloud computer activity can be checked in Atmosphere without having to bring up the VNC Viewer. - Run the bash command for searching Transdecoder-predicted proteins. As before, change the threads number and file name to match the conditions in 5.2.1.
- Run the bash command for HMMER and change the number of threads as above.
- Run the bash command for signalP and tmHMM if needed. SignalP will predict signal peptides and tmHMM predicts transmembrane protein motifs.
- Run the bash command for searching Trinity transcripts. Change the number of threads to match how many CPUs are on the instance, i.e. medium has 4 CPUs and large has 8 CPUs. Refer to step 3.1.2 for more details. Change the command Trinity.fasta to match the assembly FASTA file name.
- Loading results into the SQLite database
- Once all of the above analyses are completed, run the bash command to load output files into a final SQLite annotation database. Remove any commands for analyses that were not run.
- Export the SQLite database into a .xls file for viewing in popular table viewers.
Once the project organization files have been created (Figure 1 and 2), the first task in this workflow is to assess the raw sequencing files, and then to clean them by trimming and quality filtering. FastQC will generate human-readable summary statistics about the quality scores and length of sequences from the FASTQ file format. The FastQC figures are then compared before and after trimming to assess whether the final reads are high quality and therefore suitable for assemble. "Per base sequence quality" shows the average quality of reads across each base pair of sequencing. It is best to have a phred quality score above 20-28 indicated by the colors on the FastQC figures. "Per sequence quality score" determines whether quality filtering of reads may be necessary. If too many reads have an average score below 20-25 then it may be necessary to filter based on average read quality. "Per base sequence content" should show an even distribution across all four nucleotide bases. If there is bias in the nucleotide content is shown, then trimming ends may be necessary. "Per base GC content should also be even across all positions. If there is a wobble the reads may need to be trimmed as in 1.4.4.3. "Per sequence GC content" should be a normal distribution. Adapter or polymerase chain reaction (PCR) products can contamination in the sequencing library and skew the normal distribution. In this case, adapter trimming may be necessary. "Sequence length distribution" gives the average lengths of all reads. Reads smaller than 35-45 base pairs are usually filtered out. "Sequence duplication levels" show how many times a given read's sequence is seen within the library. Highly duplicated read sequence and count are provided in the "Overrepresented sequences" section. FastQC also attempts to identify whether the duplicated reads are adapter sequence or other known sequences associated with sequencing platforms. A label of "No Hit" means that the sequence should be investigated further using NCBI BLAST6 to determine whether it is a biologically relevant sequence, or whether it should be removed. The DE also has several versions of BLAST available. The DE BLASTn app is available at: https://de.iplantcollaborative.org/de/?type=apps&app-id=6f94cc92-6d28-45c6-aef1-036be697671d.
After raw sequencing have been screened to produce high-quality reads, the reads need to be assembled to create contiguous sequences (contigs). In brief, assemblies are created by aligning all of the short sequence reads to find similar sequences. Areas of similar sequence larger than a certain length are considered to be the same sequence because the probability of a randomly occurring similar sequence of a certain length is nearly zero. Trinity will output log files, fasta files for each step in the assembly process. However, the most important output is the final assembly file containing the contigs, which is labeled "Trinity.fasta" and found in the main folder. This file contains all of the assembled contigs, and in itself is not practically "human-readable." Therefore, the rnaQUAST tool can be used to understand the assembly in more depth. The rnaQUAST tool will output figures that will allow users to compare assemblies to determine which are most complete (Figure 4). Additional information about each figure from rnaQUAST can be found on the wiki (https://wiki.cyverse.org/wiki/x/fwuEAQ). If BUSCO7 was run, of particular interest is the specificity.txt file which shows the number of complete and partial BUSCO genes and the number of GeneMarkS-T gene predictions in an assembly. BUSCO genes are a curated sets of genes common to a group of organisms. They can be used to assess how well an assembly is capturing sets of genes that are expected to be present in any given type of organism, which is based on phylogenetic clades. A standalone BUSCO app is also available in the DE (https://de.iplantcollaborative.org/de/?type=apps&app-id=112b8a52-efd8-11e5-a15c-277125fcb1b1).
Differential gene expression analysis identifies transcripts that have different patterns of expression across treatments from simple counts per assembled transcript tables. DESeq2 uses a generalized linear model (GLM) to determine variation from a normalized mean. Experiments with replicates are preferred so that technical variation from sequencing can be normalized by the DESeq2 algorithm. DESeq2 DEG analysis yields figures and an .html report file that contains all of the output figures and a description. Alternatively, EdgeR can be used instead of DESeq2, and the same .html report will be generated with EdgeR visualizations instead. Researchers may wish to run both DESeq2 and EdgeR to find differentially expressed genes identified by both algorithms for any given experiment. Trinotate will create an output .xls file that can be opened in any spreadsheet software program. The DEG .txt files and the annotation .xls file can be analyzed and visualized in numerous downstream applications that exist outside the CyVerse platform.
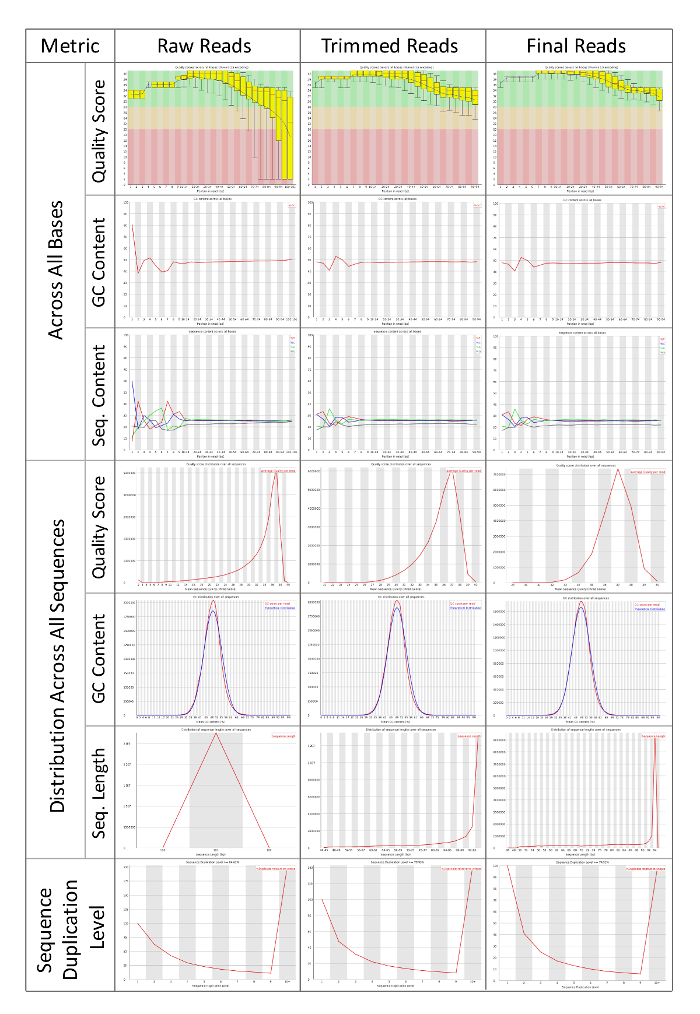
Figure 3: FastQC Reports of Raw Sequencing Reads, Trimmed Reads, and Final Trimmed and Filtered Reads. Systematic comparison of sequencing reads after each pre-processing step. High quality reads are necessary to assemble de novo transcriptomes. FastQC can help researchers to understand the initial quality of their sequencing data, and track how efficiently the reads have been pre-processed. Results from FastQC will depend the organisms and samples being sequenced, but uniformity across all samples that will be compared downstream is the primary goal of pre-processing reads. A tutorial video and documentation are available from the authors and developers of FastQC. Please click here to view a larger version of this figure.
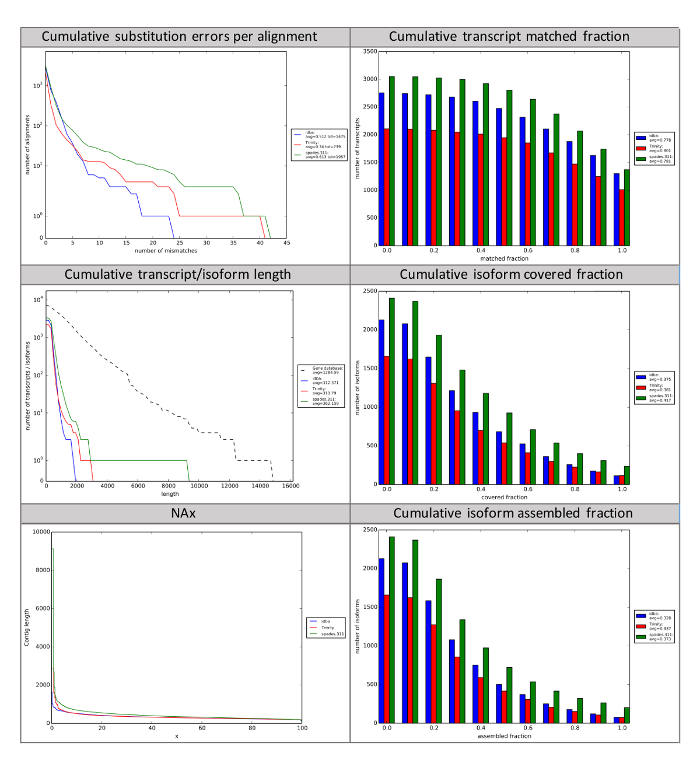
Figure 4: rnaQUAST Reports of Three Separate Assemblies. rnaQUAST can be used to compare multiple read assemblies using the same assembler, or multiple assemblers using the same initial reads. rnaQUAST leverages BUSCO to generate summary statistics about assemblies based on known core genes present in taxonomic clades. The number of mismatches per transcript and how many transcripts match to canonical genes, matched fraction, provide insight into accuracy of assemblers. The last four subplots presented here provide summary statistics of contig and isoform length and the coverage of expected isoforms. NAx represents the percentage (x) of contigs with a length longer than the length (bp) on the y-axis. Assembled fraction is the longest single assembled transcript divided by its length. Covered fraction is the percentage of complete assembled transcripts/isoforms as expected by the core prokaryotic or eukaryotic genes from BUSCO. A description of all graphs generated by rnaQUAST is available (https://wiki.cyverse.org/wiki/x/fwuEAQ). Please click here to view a larger version of this figure.
| App Name | CyVerse Platform | Third-party Documentation | CyVerse Documentation | Estimated Runtime for Sample Data Set | Link to App |
| FastQC | DE | http://www.bioinformatics. babraham.ac.uk/projects/fastqc/ https://www.youtube.com/watch?v=bz93ReOv87Y |
https://wiki.cyverse.org/wiki/pages/viewpage.action?pageId=9316768 | 15 min | https://de.iplantcollaborative. org/de/?type=apps&app-id=112b9aa8-c4a7-11e5-8209- 5f3310948295 |
| Trimmomatic v0.33 | DE | https://github.com/timflutre/trimmomatic | https://wiki.cyverse.org/wiki/display/DEapps/Trimmomatic-programmable-0.33 | 30 min | https://de.iplantcollaborative. org/de/?type=apps&app-id=9c2a30dc-028d- 11e6-a915-ab4311791e69 |
| Sickle | DE | https://github.com/najoshi/sickle | https://wiki.cyverse.org/wiki/display/DEapps/Sickle-quality-based-trimming | 30 min | https://de.iplantcollaborative. org/de/?type=apps&app-id=68b278f8-d4d6-414d-9a64-b685a7714f7c |
| Trinity | Atmosphere | https://github.com/trinityrnaseq/trinityrnaseq/wiki | https://pods.iplantcollaborative. org/wiki/display/atmman/Trinity+-+Trinotate+Atmosphere+Image |
1 week | https://atmo.iplantcollaborative. org/application/images/1261 |
| DE | https://wiki.cyverse.org/wiki/display/DEapps/Trinity-64GB-2.1.1 | 2-5 days | https://wiki.cyverse.org/wiki/display/DEapps/Trinity-64GB-2.1.1 | ||
| rnaQUAST v1.2.0 | DE, Atmosphere | http://spades.bioinf.spbau.ru/rnaquast/release1.2.0/manual.html | https://pods.iplantcollaborative. org/wiki/display/TUT/rnaQUAST+1.2.0+%28denovo+based%29+using+DE |
30 min | https://de.iplantcollaborative. org/de/?type=apps&app-id=980dd11a-1666- 11e6-9122-930 ba8f23352 |
| Transdecoder | DE | https://transdecoder.github.io | https://wiki.cyverse.org/wiki/display/DEapps/Transcript+decoder+2.0 | 2-3 hours | https://de.iplantcollaborative. org/de/?type=apps&app-id=5a0ba87e-b0fa-4994-92a2- 0d48ee881179 |
| DESeq2 | DE | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | https://pods.iplantcollaborative. org/wiki/pages/viewpage.action?pageId=28115142 |
2-3 hours | https://de.iplantcollaborative. org/de/?type=apps&app-id=9574e87c-4f90- 11e6-a594-008 cfa5ae621 |
| EdgeR | DE | https://bioconductor.org/packages/release/bioc/vignettes/edgeR/inst/doc/edgeR.pdf | https://wiki.cyverse.org/wiki/pages/viewpage.action?pageId=28115144 | 2-3 hours | https://de.iplantcollaborative. org/de/?type=apps&app-id=4a08ceda-54fe- 11e6-862f-008 cfa5ae621 |
| Trinotate | Atmosphere | https://trinotate.github.io/ | https://pods.iplantcollaborative. org/wiki/display/atmman/Trinity+-+Trinotate+Atmosphere+Image |
1 week | https://atmo.iplantcollaborative. org/application/images/1261 |
Table 1: Analysis Programs, Platforms they are Available on, and Additional Resources Available for the Workflows in Order by First Appearance. All Package versions are current as of April 2016.
