Investigation of Activated Mouse Olfactory Sensory Neurons via Combined Immunostaining and in situ Hybridization
Summary
This protocol combines in situ hybridization with immunofluorescence to identify olfactory and vomeronasal receptor genes expressed in olfactory sensory neurons after activation by chemical stimuli in the mouse.
Abstract
Animals rely on chemical communication to convey and perceive relevant environmental information, ranging from assessment of food quality to detection of available mating partners or threats. In mice, this task is executed primarily by the olfactory system and its underlying subsystems, including the main and accessory olfactory systems. Both have peripheral organs populated by sensory neurons expressing G-protein coupled receptors able to bind chemical cues that reach the nasal cavity. Even though the molecular characteristics of these receptors is well understood, little is known about their cognate specific ligands. The method described here combines in situ hybridization detection of olfactory or vomeronasal receptors with immunodetection of phosphorylated ribosomal protein S6 (pS6) – a marker of neuronal activation. This protocol was devised to identify neurons activated after a single event of exposure to purified or complex chemical stimuli detected by the olfactory organs. Importantly, this technique allows the investigation of neurons triggered in biologically relevant contexts. Ideally, this method should be used to probe the molecular biology of the olfactory system and to study olfactory behaviors.
Introduction
Chemosignaling is the most widespread form of communication in animals. In mammals, detection of chemical cues is mediated mainly through olfaction and is paramount for finding food, locating possible mating partners, and avoiding potential predators1,2,3. The mouse olfactory system is further divided into different subsystems, each with their own anatomical, physiological and molecular properties4. Among these, the main olfactory system (MOS) and accessory olfactory system (AOS) are the most widely studied and better characterized.
The MOS is mostly responsible for the detection of volatile chemical molecules that reach the nasal cavity. This is accomplished by olfactory sensory neurons (OSNs), which populate the system's sensory interface – the main olfactory epithelium (MOE). Each OSN expresses one out of thousands of olfactory receptors (ORs) in a monogenic and monoallelic fashion5. Furthermore, ORs tend to be broadly tuned6, and a single odorant may bind to more than one OR, generating a combinatorial code for odors7.
In turn, the AOS is mainly responsible for pheromone and kairomone detection. These ligands activate vomeronasal sensory neuros (VSNs) in the vomeronasal organ (VNO). VSNs in the apical zone of the VNO express receptors from the vomeronasal receptor 1 family (V1Rs)8, while neurons in the basal zone express the vomeronasal receptor 2 family (V2Rs)9. Importantly, it has been shown that some VSNs co-express more than one vomeronasal receptor10,11,12.
Despite all available information on the molecular characteristics of ORs and VRs, knowledge on their cognate ligands is still very limited. The identification of specific olfactory neurons responsible for the detection of a molecule or complex stimulus is an important task for those investigating olfaction, olfactory-driven behaviors, and physiological responses. Several strategies have been attempted to deorphanize olfactory receptors, including calcium-imaging7, single-cell RNA sequencing13,14, heterologous receptor expression15, and strategies based on immediate early genes (IEGs)16. More recently, some techniques have been employed to assess the modulatory effects of odorants in sensory neurons, using in vivo calcium imaging17,18 and fluorescent-protein tagged single-cell RNA sequencing strategies19. Most of these techniques demand an artificial setup or are performed on dissociated neurons, making it impossible to simultaneously assess behaviors triggered by the odors used.
The protocol described here allows the identification of sensory neurons activated after a single event of odor exposure, by combining riboprobes that detect specific ORs or VRs with the immunodetection of phosphorylated S6 ribosomal protein (pS6), a proxy for neuronal activation. This method is a reliable way to investigate the identity of sensory neurons activated by different chemosignals in a biologically relevant context.
Protocol
Animal procedures were carried out in accordance with Animal Protocol #1883-1, approved by the University of Campinas (Institute of Biology's Institutional Animal Care and Use Committee – Committee for Ethics in Animal Use in Research), which follows the guidelines established by the federal National Council for Animal Experimentation Control (CONCEA).
1. Material preparation
- Prepare 1 L of 3% H2O2 (v/v) in 1x PBS. Dilute 100 mL 10× PBS and 100 mL of 30% H2O2 in 500 mL of ultrapure water. Complete the volume with ultrapure water. Prepare this solution immediately before use.
- Prepare a humidified chamber containing pieces of lint-free laboratory wipes wet with 5× SSC solution.
- Prepare hybridization wash solutions: 5× SSC (at 55 °C), 2× SSC (55 °C), 0.2× SSC (55 °C) and 0.1× SSC (55 °C and room temperature). Dissolve the appropriate volumes of 20× SSC stock solution in RNase-free ultrapure water. Prepare the diluted solutions fresh before the start of each experiment.
- Prepare immunohistochemistry blocking solution: 1× PBS/1% BSA/0.1% Triton X-100. For 100 mL, dilute 10 mL of 10× PBS, 10 mL of 10% BSA and 3.33 mL of 3% Triton X-100 in 70 mL of ultrapure water. Complete the volume with ultrapure water. Prepare the diluted solutions fresh before the start of each experiment.
- Prepare PTw solution: 1× PBS/0.1% Tween-20. For 100 mL, dilute 1 mL of 10% Tween-20 and 10 mL of 10× PBS in 70 mL of ultrapure water. Complete the volume with ultrapure water. Prepare this solution fresh before the start of each experiment.
- Prepare RNase-free 0.1 M triethanolamine solution. For 250 mL, dilute 3.33 mL of reagent-grade triethanolamine in 200 mL of RNase-free ultrapure water under agitation (use RNase-free glassware and magnetic stirrer when preparing this solution). Adjust pH to 8.0 with HCl. Complete the volume with RNase-free ultrapure water and store protected from light until use. Prepare this solution fresh before the start of each experiment.
- Prepare RNase-free 0.1% H2O2 (v/v) in 1× PBS. For 1 L, dilute 100 mL of 10× PBS and 3.3 mL of 30% H2O2 in 500 mL of RNase-free ultrapure water. Complete the volume with RNase-free ultrapure water. Prepare this solution immediately before use.
- Prepare RNase-free solutions of the following: 0.2 M HCl; 1 M Tris-HCl pH 8.0; 10 mg/mL yeast tRNA; 10% SDS; 10× PBS; 100× Denhardt's solution; 20× SSC; 50% dextran sulfate; 6 M HCl
- Prepare RNase-free 4% paraformaldehyde fixative solution. For 100 mL, dissolve 4 g of paraformaldehyde in 80 mL of 1× PBS. Adjust pH to 7.4 with 10 M NaOH. Complete the volume with 1× PBS. This solution must be prepared fresh before the start of each experiment.
Caution: Paraformaldehyde is a hazardous substance that can potentially cause harm if inhaled. Prepare solution under a fume hood and wear protective gloves, mask, and safety goggles. - Prepare RNase-free dissection solution: 30% sucrose/0.45 M EDTA pH 8.0/1× PBS. For 100 mL, dissolve 30 g of sucrose in 60 mL of RNase-free 0.5 M EDTA pH 8.0. Add 10 mL of 10× PBS. Complete the volume with RNase-free 0.5 M EDTA pH 8.0.
- Prepare RNase-free glass or plastic graduated cylinders and beakers.
- Prepare RNase-free pre-hybridization and hybridization solutions: 50% deionized formamide (v/v)/600 mM NaCl/200 μg/mL yeast tRNA/0.25% SDS (w/v)/10 mM Tris-HCl pH 8.0/1× Denhardt's solution/1 mM EDTA pH 8.0/10% dextran sulfate (w/v). For 10 mL in a graduated cylinder, add 5 mL of deionized formamide, 1.2 mL of 5 M NaCl, 200 μL of 10 mg/mL yeast tRNA, 250 μL of 10% SDS, 100μL of 1 M Tris-HCl pH 8.0, 200 μL of 50× Denhardt's solution, 20μL of 500 mM EDTA pH 8.0, and 2 mL of 50% dextran sulfate (M.W. 500,000). Complete the volume with RNase-free ultrapure water. Prepare this solution fresh before the start of each experiment.
NOTE: Dextran sulfate is very viscous. It is advisable to prepare an extra 20% of the total volume of hybridization/pre-hybridization solutions to account for pipetting errors. Yeast tRNA may be omitted from the pre-hybridization solution, if desired. - Prepare thermoplastic laboratory film coverslips. Cut rectangular thermoplastic laboratory film pieces 1-2 mm shorter than the length and width of a microscope slide.
- Prepare TN buffer: 100 mM Tris-HCl/150 mM NaCl. For 100 mL, dilute 10 mL of 1 M Tris-HCl pH 7.5 and 3 mL of 5 M NaCl in 70 mL of ultrapure water. Complete the volume with ultrapure water. Prepare this solution fresh before the start of each experiment.
- Prepare TNB blocking buffer: 0.5% blocking reagent A (w/v) in TN Buffer. For 200 mL, dissolve 1 g of blocking reagent A in 200 mL of TN Buffer (see Table of Materials for commercial source of blocking reagent A).
- Prepare TNT buffer: 0.05% Tween-20 in TN Buffer. For 1 L, dilute 5 mL of 10% Tween-20 in 950 mL of TN buffer. Slowly complete the volume with TN buffer to avoid foaming. Prepare this solution fresh before the start of each experiment.
- Prepare Tyramide-Alexa 488 working solution: 1× amplification buffer A/0.0015% H2O2/1:100 tyramide-Alexa 488. For 100 μL, dilute 1 μL of 0.15% H2O2 and 1 μL of tyramide-Alexa 488 in 98 μL of amplification buffer A (see Table of Materials for commercial sources of tyramide-Alexa 488 and amplification buffer A). Prepare this solution immediately before use.
- Prepare Tyramide-Alexa 555 working solution: 1× amplification buffer A/0.0015% H2O2/1:100 tyramide-Alexa 555. For 100 μL, dilute 1 μL of 0.15% H2O2 and 1 μL of tyramide-Alexa 555 in 98 μL of amplification buffer A (see Table of Materials for commercial sources of tyramide-Alexa 555 and amplification buffer A). Prepare this solution immediately before use.
- Prepare Tyramide-biotin working solution: 1× amplification buffer B/0.0015% H2O2/1:50 tyramide-biotin. For 100 μL, dilute 1 μL of 0.15% H2O2 and 2 μL of tyramide-biotin in 97 μL of amplification buffer B (see Table of Materials for commercial sources of tyramide-biotin and amplification buffer B). Prepare this solution immediately before use.
- Bake glassware at 200 °C for at least 4 h. Treat non-disposable plasticware (including slide racks, Coplin jars and syringes) with 3% H2O2 for 15-30 min, followed by thorough washing under RNase-free water. Treat stainless steel metal instruments, such as dissection tools, forceps, and cryostat specimen holders, with 3% H2O2 for 15 min, followed by thorough washing under RNase-free water. Clean bench surfaces, heated plate, and dry blocks with RNase cleaning solution.
2. Riboprobe synthesis
NOTE: The following procedure to synthesize 1 kb digoxigenin-labeled cRNA probes for in situ hybridization via in vitro transcription is based on previously published protocols20,21,22,23.
- Use appropriate oligonucleotide primer pairs to amplify a 1 kb DNA fragment from the gene of interest (e.g., olfactory receptor gene). Select primer pairs that amplify a region present in the mature mRNA to be detected (for example, the gene's coding sequence). A selection of primers for different olfactory receptors is provided in the Table of Materials. For details on probes to detect other vomeronasal olfactory receptors, please check previous publications16,21.
- Run the PCR products on an 0.8% agarose gel and remove the desired 1 kb band.
- Purify the DNA fragment using a suitable mini column-based gel purification kit according to manufacturer's protocol.
- Quantify the purified DNA fragment and clone it into a suitable PCR cloning vector according to manufacturer's protocol. It is advisable to use cloning vectors containing T7 and SP6 RNA polymerase promoters flanking the cloned amplicon.
- Transform the recombinant plasmid into competent recombination-free bacteria via chemical transformation or electroporation according to manufacturer's protocol.
- Seed isolated transformant bacterial colonies into rich medium and isolate the plasmid via DNA mini-preparation according to manufacturer's protocol.
- Perform restriction digestion analysis with suitable restriction enzymes and/or Sanger sequencing to check the identity and orientation of the inserted DNA fragment according to manufacturer's protocol.
- Perform restriction digestion to linearize 10 μg of the plasmid using an appropriate restriction enzyme. To produce an anti-sense riboprobe after in vitro transcription, use a restriction enzyme that digests the plasmid DNA at the extremity of the insert opposite to the RNA polymerase promoter located downstream to the gene's coding sequence. To produce sense riboprobes after in vitro transcription, use a restriction enzyme that digests the plasmid DNA at the extremity of the insert opposite to the RNA polymerase promoter located upstream to the gene's coding sequence.
- Run an aliquot of the plasmid linearization reaction product on a 0.8% agarose gel to confirm completeness of digestion. Once complete linearization is confirmed, purify the remainder of the reaction using a column-based PCR cleanup kit according to manufacturer's protocol.
- Synthesize digoxigenin-labeled cRNA probes by in vitro transcription using 1-2 μg of the linearized plasmid as a template, the appropriate enzyme (e.g., T7 or SP6 RNA polymerase), and a suitable digoxigenin (DIG) labeling kit.
- Remove the DNA template from the reaction by digesting with DNase I at 37 °C for 30 min.
- Purify the cRNA probes using either a mini column-based RNA purification kit or a gel filtration-based purification kit. Elute the resulting probe in 50 μL of RNase-free water.
- Quantify the resulting riboprobe, preferably using fluorometric methods. Usually, the expected yield is above 100 ng/μL.
- Run the riboprobe on an automated electrophoresis system or on an RNase-free 0.8% agarose gel to check for probe quality and degradation.
- Store the synthesized probe at -80 °C until needed.
3. Exposure of mice to olfactory stimuli
- Individually house each animal in a clean cage for at least 24 h prior to exposure. Remove the food grid at least 2 h before exposure. All animals used in this study were 8-12 weeks old male C57BL/6 mice (average weight: 20 g).
- Prepare the appropriate olfactory stimulus. Different stimuli can be used to activate olfactory neurons in the MOE (main olfactory epithelium) or VNO (vomeronasal organ). For a comprehensive list of olfactory stimuli, please consult other publications3, 16, 22,23,24. Liquid stimuli, such as chemical solutions, urine, or recombinant protein solution, can be deposited on a medical gauze or cotton ball.
- Insert the olfactory stimulus into the cage and leave the animal undisturbed for 1 h.
- Euthanize the mouse subject and quickly dissect the MOE or VNO using RNase-free dissecting tools.
4. Tissue dissection and freezing
- Dissect the MOE or VNO under a stereomicroscope using dissection solution.
- For the VNO, remove the thick septal bones, leaving the softer cartilage shells surrounding the organ.
- Prepare specimen for cryo-sectioning.
- Blot the specimen dry on a piece of blotting paper and place it inside a histological plastic mold containing embedding medium.
- For VNO, arrange each organ vertically and pull it to the bottom of the plastic mold with the help of a 1 mL syringe (Figure 1A).
- For MOE, place it at the bottom of the plastic mold with the cribriform plate facing down.
- Remove the excess of bubbles around the specimen since these can interfere with sectioning.
- Freeze specimens on dry ice.
- Store the specimen block at -80 °C.
NOTE: Frozen specimens may be stored for several months before sectioning.
- Perform cryo-sectioning.
- Turn the cryostat on and set the temperature to -23 °C at least 4 h prior to use.
- Remove the frozen block from the histological plastic mold and trim it with a razor blade.
- Leave 5 mm around the specimen, as this makes section handling easier (Figure 1B).
- Use embedding medium to attach the block to the cryostat's specimen holder.
- Collect 16 μm sections onto positively charged microscope slides.
- Store the slides at -80 °C.
NOTE: Slides may be stored at -80 °C for several months before in situ hybridization.
5. Step-by-step in situ hybridization protocol
- Dry the slides for 10 min using a hair dryer. Use the warm jet to defrost the slides, then switch to room-temperature jet until the embedding medium is opaque.
- Add 500 μL per slide of RNase-free 4% paraformaldehyde fixative solution and incubate at 23-26 °C for 15 min to fix the histological sections.
- Wash the slides twice in RNase-free 1× PBS for 5 min each.
- Add 500 μL per slide of RNase-free 0.2 M HCl and incubate at 23-26 °C for 10 min.
- Wash the slides twice in RNase-free 1× PBS for 5 min each.
- Add 500 μL per slide of RNase-free 0.1% H2O2/1× PBS and incubate for 30 min at 23-26 °C. Perform this step in the dark to prevent light-induced degradation of hydrogen peroxide.
- Wash slides twice in RNase-free 1× PBS for 5 min each.
- Perform acetylation by transferring slides to a jar containing 250 mL of RNase-free triethanolamine 0.1 M pH 8.0 and a magnetic stir bar, in a fume hood. Add 625 μL of acetic anhydride under constant stirring. Turn stirring speed down, add another 625 μL drop by drop onto the slides, then turn stirring off and incubate for 10 min.
- Wash slides twice in RNase-free 1× PBS for 5 min each.
- Wash slides once in RNase-free 1× PBS at 60 °C for 5 min.
- Perform pre-hybridization by removing slides from 1× PBS one at a time and blotting each of them dry using lint-free laboratory tissue paper. Pipet 400 μL of pre-hybridization solution onto each slide and place it inside a humidified chamber pre-warmed at 60 °C in a water bath or heated oven. Incubate at 60 °C for 1 h.
- Heat up enough hybridization solution to 60 °C, aliquot 200 μL per slide in microfuge tubes, and heat them up to 85 °C for 10 min.
- Add the appropriate cRNA probe(s) to the hybridization solution tubes (1 μg/mL each probe) and denature at 85 °C for 5 min.
- For hybridization, take one slide at a time from the humidified chamber, blot it dry and place it onto a 60 °C heated plate. Add 200 μL of riboprobe-containing hybridization solution and cover the slide with a glass coverslip. Incubate for 12-16 h in the humidified chamber at 60 °C.
NOTE: Do not allow slides to cool down after pre-hybridization. Hybridization temperature must be empirically determined for each probe. Sixty degrees Celsius is suggested as a starting point, since it generally works well for 1 kb probes. - Heat SSC wash solutions to 55 °C in separate jars and place them in a water bath at the appropriate temperature.
- After hybridization is finished, remove the coverslip from each slide using a bath containing 5× SSC solution at 55 °C. Hold each slide horizontally inside the warm SSC solution. When the coverslip has detached, tilt the slide and let the coverslip fall down to the bottom.
- Wash with 2× SSC at 55 °C for 30 min in a slide jar.
- Wash with 0.2× SSC at 55 °C for 20 min in a slide jar.
- Wash with 0.1× SSC at 55 °C for 20 min in a slide jar.
- Transfer to 0.1× SSC at 23-26 °C and incubate for 3 min in a slide jar.
- Wash in PTw for 10 min in a slide jar.
- Wash twice in TN Buffer, for 5 min each, in a slide jar.
- Pipet 600 µL of TNB blocking buffer onto each slide and incubate for 3 h at 23-26 °C.
- Pipet 500 µL of primary antibody solution (peroxidase-conjugated anti-DIG antibody diluted 1:400 in TNB solution) onto each slide. Carefully overlay a piece of thermoplastic laboratory film onto each slide and incubate for 12-16 h at 4 °C.
- Wash slides 6 times with TNT Buffer for 5 min each, under mild stirring in a slide jar.
- Pipet 100 µL of tyramide-biotin working solution onto each slide. Cover with thermoplastic laboratory film coverslips and incubate for 12 min at 23-26 °C.
NOTE: Perform this incubation step in a staggered fashion, with 2 or 3 slides per batch, to allow good control over incubation times. Duration of tyramide incubation may need to be optimized, depending on cRNA probe sensitivity and concentration. - Wash slides 6 times with TNT Buffer for 5 min each, under mild stirring in a slide jar.
- Pipet 200 µL of solution containing peroxidase-conjugated streptavidin (SA-HRP, diluted 1:100 in TNB buffer) onto each slide and cover with a thermoplastic laboratory film coverslip. Incubate at 23-26 °C for 1 h.
- Wash slides 6 times with TNT Buffer for 5 min each, under mild stirring in a slide jar.
- Pipet 100 μL of tyramide-Alexa 488 working solution onto each slide, cover with thermoplastic laboratory film coverslip and incubate at 23-26 °C for 12 min in the dark.
- Wash slides 6 times with TNT Buffer for 5 min each, under mild stirring in a slide jar.
- Incubate in 3% H2O2/1× PBS in a slide jar for 1 h, to inactivate peroxidases from the previous steps.
- Wash slides 6 times with TNT Buffer for 5 min each, under mild stirring in a slide jar.
6. pS6 immunostaining
- Pipet 500 μL of 4% paraformaldehyde/1× PBS solution onto each slide and incubate at 23-26 °C for 15 min to fix sections.
- Pipet 500 μL of 1× PBS/0.1% Triton X-100 onto each slide and incubate for 5 min to permeabilize sections.
- Wash twice with 1× PBS in a slide jar.
- Pipet 2-3 drops per section of commercially available tyramide signal amplification blocking solution B (Table of Materials) and incubate at 23-26 °C for 1 h.
- Pipet 400 μL of immunohistochemistry blocking solution onto each slide and incubate at 23-26 °C for 30 min to block sections.
- Pipet 200 μL of rabbit anti-pS6 primary antibody solution (diluted 1:200 in immunohistochemistry blocking solution; final concentration: 1 μg/mL) onto each slide, cover with thermoplastic laboratory film coverslip, and incubate at 4 °C for 12-16 h.
- Wash 3 times with 1× PBS/0.1% Triton X-100, for 5 min each in a slide jar.
- Pipet 2-3 drops per slide of commercially available peroxidase-conjugated anti-rabbit secondary antibody solution (Table of Materials) and incubate at 23-26 °C for 1.5 h.
- Wash 3 times with 1× PBS/0.1% Triton X-100, for 5 min each in a slide jar.
- Blot slides dry and pipet 100 to 200 μL of tyramide-Alexa 555 working solution onto each slide, overlay with a piece of thermoplastic laboratory film coverslip, and incubate for 7 min in the dark.
- Wash twice with 1× PBS for 5 min each in a slide jar.
- Pipet 500 μL of diluted nuclear stain (in 1× PBS) onto each slide and incubate at 23-26 °C for 30 min.
- Wash twice with 1× PBS at 23-26 °C for 5 min each in a slide jar.
- Mount slides with anti-fading mounting medium and let the mounting medium cure for 24h in the dark.
- Store slides at the appropriate temperature until imaging, to avoid fluorescence fading.
7. Microscopy imaging
- Image slides using a epifluorescence or confocal microscope equipped with appropriate filters for the fluorophores used.
Representative Results
The current protocol aims at obtaining microscopy images in which the experimenter's gene of interest and the neuronal activity marker pS6 are fluorescently labeled. The described method involves Tyramide Signal Amplification, which produces clear and strong labeling with little to no background. pS6 immunostaining appears as cytoplasmic fluorescent signal that usually fills the entire neuronal cell body, whereas in situ hybridization signal for olfactory receptor neurons shows as cytoplasmic staining. pS6 is a transient marker of olfactory neuron activation and therefore the associated immunostaining signal will appear stronger in cells that have been activated in the 1 h period preceding sensory organ fixation. Fluorescence crosstalk between staining for pS6 and receptors is not expected, provided the appropriate bandpass microscopy filters are used.
Labeled sections can be used to evaluate co-localization between the two types of staining, allowing for the identification of genes expressed by the population of activated neurons under study. Figure 2A,B shows VNO images from a mouse exposed to cat odor, with co-localization of signals for V2R receptors in clade A4 and for pS6, indicating that neurons expressing those olfactory receptors are activated by the stimulus used. Conversely, when activated neurons do not express the olfactory receptors under scrutiny, no co-localization between signals is expected (Figure 2C,D).
Challenges inherent to this protocol include maintaining the integrity of cryostat sections, particularly because the olfactory organs are dissected without fixation. This may result in torn sections (Figure 3A) or partially deformed or curled sections during subsequent incubations (Figure 3B). Another potential problem is high fluorescence background staining, possibly derived from insufficient blocking or excessive signal development in amplification steps, in both pS6 immunohistochemistry (Figure 3C) or in situ hybridization steps (Figure 3D).
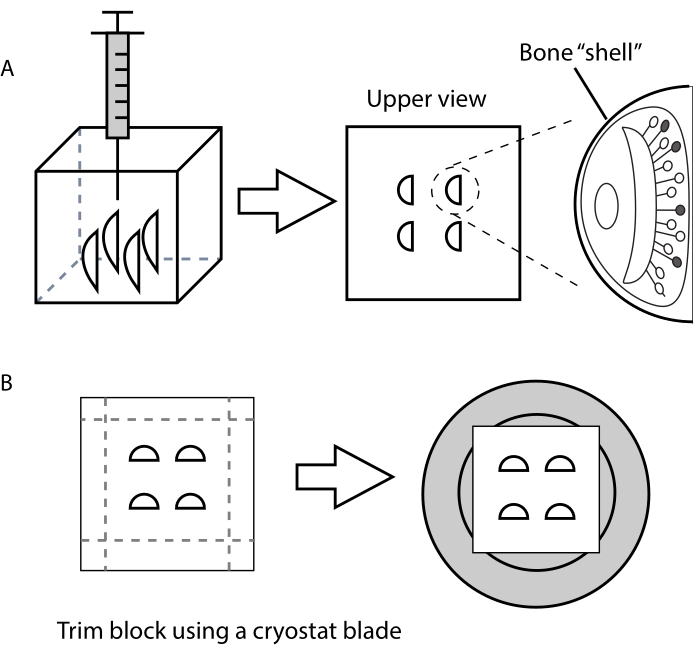
Figure 1. Schematic representation showing vertical placement of the VNOs inside the embedding medium to obtain transversal cryostat sections. (A) Use of a syringe to place the VNOs inside the histology mold, with the bone shell facing sideways. (B) Instructions for trimming the specimen block and attaching it to the cryostat holder (grey). Please click here to view a larger version of this figure.
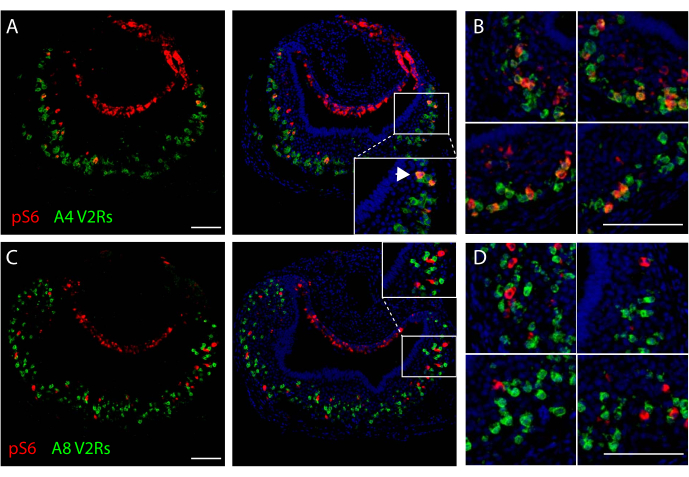
Figure 2. Representative images of dual in situ hybridization/pS6 immunohistochemistry staining of VNOs after mouse exposure to cat odors. (A) Microscopy image after in situ hybridization using riboprobes for V2R receptors in clade A4 (green fluorescence) and pS6 immunostaining (red). Middle panel: overlay of dual staining and nuclear counterstaining (blue). Arrowheads indicate co-localization of in situ hybridization and pS6 immunohistochemistry signals. (B) High magnification images from different VNO sections subjected to dual staining in A. (C) Microscopy image after in situ hybridization using probes for V2R receptors in clade A8 (green fluorescence) and pS6 immunohistochemistry (red). Middle panel: overlay of dual staining and nuclear counterstaining (blue). (D) High magnification images from different VNO sections subjected to the same dual staining as in C. Scale bars are 100 μm. Please click here to view a larger version of this figure.

Figure 3. Representative suboptimal results. (A) Damaged, teared VNO (arrow). (B) Curled VNO section (arrow). (C) High background in pS6 immunostaining signal. Arrow shows an example of border effect. (D) High background staining after riboprobe in situ detection. Arrowheads indicate true staining. Scale bars are 100 μm. Please click here to view a larger version of this figure.
| Problem | Possible causes | Proposed solution |
| Tears in sections or difficulty when sectioning | Bones are impairing sectioning process | Replace cryostat blade or change cryostat block orientation until section quality improves. Usually, the remaining bones, if present, are positioned opposite to the side where the cryostat blade first hits the specimen. |
| Loss of sections during staining | Use of non-adhesive microscope slides | Use silanized or other type of positively-charged slides. Consider changing brands. |
| Harsh conditions in situ hyb steps | Handle slides carefully during staining washes, especially when inserting and removing from wash solutions and during coverslip displacement. | |
| Low in situ hybridization signal | Insufficient probe hybridization | Increase probe concentration. Reduce hybridization or probe wash temperature. |
| Poor tyramide signal ampification | Extend duration of tyramide amplification steps. | |
| High staining background | Insufficient blocking | Check blocking solution concentration or extend blocking duration. |
| Excessive tyramide signal ampification | Reduce duration of tyramide amplification steps. | |
| Sections drying up during staining | Maintain sections covered in solution at all times. Minimize time between washes, during which slides are outside the staining jars and may dry up. |
Table 1. Troubleshooting table with the most frequent problems and proposed solutions. This table presents steps to troubleshoot the protocol, in cases where tears are found in sections or when there is difficulty sectioning. Other problems are also addressed, such as loss of sections during staining, low in situ hybridization signal, and high staining background.
Discussion
The protocol described here reliably identifies sensory neurons activated by chemical cues in the olfactory system through a combination of in situ detection of OR or VR receptors with immunodetection of pS6, an indirect marker of neuronal activity. The experimenter must take extra caution to maintain histology integrity and perform all steps before hybridization under RNase-free conditions. Failure to do so may cause mRNA degradation and compromise riboprobe labeling. Care must be exercised during dissection of the olfactory organs, as unfixed tissue is particularly fragile and may be easily damaged.
In situ hybridization in this protocol involves two rounds of signal amplification, and it must be noted that fluorescence intensity may be affected by factors related to hybridization or signal development. For example, incubation temperatures and periods must be empirically optimized. Weak labeling may be due to low probe concentration, high hybridization temperature, or short tyramide labeling. Conversely, high background may be due to low hybridization temperature or over-development in tyramide incubation steps. These parameters must be adjusted to attain optimal signal-to-background ratio (see Table 1 for a thorough troubleshooting discussion).
Similarly, pS6 immunodetection may lead to low fluorescence intensity or high background staining. These problems generally arise from inadequate duration of tyramide incubation steps, insufficient blocking, or inadequate quenching of endogenous peroxidases (Table 1).
Despite such methodological concerns, the procedure described here has important advantages. pS6 immunodetection is simpler and faster than double in situ hybridization with riboprobes designed to target popular IEGs, such as c-fos, Arc, and Egr-1. The method described here generally produces very clear labeling in olfactory sensory neurons23,25,26. Furthermore, the technique allows the identification of activated neurons in biologically relevant contexts (i.e., upon the detection of odors from a potential mating partner or from a predator), in experimental paradigms where behaviors can be simultaneously assessed and recorded. Other types of investigation aiming to detect olfactory neuron activation do not share this advantage, as they employ artificial settings in which the animal is head-fixed or euthanized prior to neural recordings or calcium imaging. The ability to identify neurons activated by specific chemical cues in freely moving mice is a powerful asset in several research areas, ranging from those studying the molecular biology of the olfactory system and chemical ecology to those interested in behavioral neuroscience and ethology. This protocol provides a reliable method for the investigation of chemodetection in these studies.
Declarações
The authors have nothing to disclose.
Acknowledgements
We thank GAG Pereira and JA Yunes for resources, APF Ferreira and WO Bragança for administrative and technical help, and the Life Sciences Core Facility (LaCTAD-UNICAMP) staff for help with confocal microscopy. This work was supported by the Sao Paulo Research Foundation (FAPESP; grant numbers 2009/00473-0 and 2015/50371-0 to F.P.), by PRP/UNICAMP (grant numbers 2969/16, 725/15, 348/14, and 315/12 to F.P.), by FAPESP fellowships to V.M.A.C. (2014/25594-3, 2012/21786-0, 2012/01689-0), T.S.N. (2012/04026-1), and by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) fellowship to T.S.N.
Materials
| 10x phosphate-buffered saline (PBS) | Thermo Fisher Scientific | AM9625 | n/a |
| 20x amplification diluent (reaction buffer from Alexa Fluor 555 Tyramide SuperBoost kit) | Thermo Fisher Scientific | B40923 or B40922 | refered to as Amplification Buffer A in working solutions for tyramide-Alexa 488 or tyramide-Alexa 555 signal amplification |
| 20x SSC | Merck (Calbiochem) | 8310-OP | n/a |
| 30% Bovine Serum Albumin (BSA) | Merck (Sigma-Aldrich) | A9576 | n/a |
| 30% hydrogen peroxyde (H2O2) | Merck (Sigma-Aldrich) | H1009 | n/a |
| Acetic anhydride | Merck (Sigma-Aldrich) | 320102 | n/a |
| Agarose | Merck (Sigma-Aldrich) | A9539 | n/a |
| Amplification diluent (from TSA Biotin kit) | Akoya Biosciences (Perkin Elmer) | SAT700001EA | refered to as Amplification Buffer B in working solution for tyramide-biotin signal amplification |
| Anti-pS6 (Ser 244/247) rabbit polyclonal antibody | Thermo Fisher Scientific | Cat# 44-923G, RRID:AB_2533798 | n/a |
| Blocking buffer 1x (from Alexa Fluor 488 Tyramide SuperBoost kit) | Thermo Fisher Scientific | B40923 or B40922 | refered to as blocking solution B in the tyramide signal development step |
| Blocking reagent (from TSA Biotin kit) | Akoya Biosciences (Perkin Elmer) | SAT700001EA | refered to as blocking reagent A in the formulation for TNB buffer |
| DAPI nuclear stain | Thermo Fisher Scientific | D1306 | n/a |
| Denhardt's solution (50x) | Merck (Sigma-Aldrich) | D9905 | n/a |
| Deoinized formamide | Merck (Sigma-Aldrich) | F9037 | n/a |
| Dextran sulfate solution (50%) | Merck (Chemicon) | S4030 | n/a |
| Ethylene-diamine-tetraacetic acid (EDTA) | Merck (Sigma-Aldrich) | E9884 | n/a |
| Hoechst 33342 nuclear stain | Thermo Fisher Scientific | H1399 | n/a |
| Hydrochloric acid (HCl) | Merck (Sigma-Aldrich) | 320331 | n/a |
| Olfactory stimuli | n/a | Papes et al. (2010), Carvalho et al. (2015), Nakahara er al. (2016), Carvalho et al. (2020) | n/a |
| Paraformaldehyde | Merck (Sigma-Aldrich) | P6148 | n/a |
| Peroxidase-conjugated anti-digoxigenin antibody (Fab fragments) | Merck (Roche) | 11207733910; RRID:AB_514500 | refered to as peroxidase-conjugated anti-DIG antibody |
| Peroxidase-conjugated anti-rabbit secondary antibody (polyHRP-conjugated goat anti-rabbit reagent from Alexa Fluor 488 Tyramide SuperBoost kit) | Thermo Fisher Scientific | B40922 | n/a |
| Peroxidase-conjugated streptavidin (from TSA biotin kit) | Akoya Biosciences (Perkin Elmer) | SAT700001EA | n/a |
| ProLong Gold antifade mountant | Thermo Fisher Scientific | P36934 | refered to as anti-fading mounting medium |
| RNase-free ultrapure water | Thermo Fisher Scientific | 10977015 | n/a |
| Sodium chloride (NaCl) | Merck (Sigma-Aldrich) | S9888 | n/a |
| Sodium dodecyl sulfate (SDS) | Merck (Sigma-Aldrich) | 436143 | n/a |
| Sodium hydroxide (NaOH) | Merck (Sigma-Aldrich) | S8045 | n/a |
| Sucrose | Merck (Sigma-Aldrich) | S0389 | n/a |
| Triethanolamine | Merck (Sigma-Aldrich) | T58300 | n/a |
| Triton X-100 | Merck (Sigma-Aldrich) | X100 | n/a |
| Trizma hydrochloride (Tris-Cl) | Merck (Sigma-Aldrich) | T5941 | n/a |
| Tween-20 | Merck (Sigma-Aldrich) | 822184 | n/a |
| Tyramide-Alexa 488 conjugate (from Alexa Fluor 488 Tyramide SuperBoost kit) | Thermo Fisher Scientific | B40922 | n/a |
| Tyramide-Alexa 555 conjugate (from Alexa Fluor 555 Tyramide SuperBoost kit) | Thermo Fisher Scientific | B40923 | n/a |
| Tyramide-Biotin conjugate (from TSA Biotin kit) | Akoya Biosciences (Perkin Elmer) | SAT700001EA | n/a |
| Yeast tRNA | Merck (Roche) | 10109495001 | n/a |
| Critical Commercial Assays and Animals | |||
| Alexa Fluor 488 Tyramide SuperBoost kit | Thermo Fisher Scientific | B40922 | n/a |
| Alexa Fluor 555 Tyramide SuperBoost kit | Thermo Fisher Scientific | B40923 | n/a |
| DIG RNA Labeling Kit (SP6/T7) | Merck (Roche) | 11175025910 | n/a |
| High Sensitivity RNA ScreenTape Analysis reagents (buffer, ladder, and tape) | Agilent | 5067-5580, 5067-5581, and 5067-5579 | refered to as automated electrophoresis system |
| Mouse: C57BL/6J inbred strain | Jackson Laboratories | Stock No: 000664; RRID:IMSR_JAX:000664) | n/a |
| ProbeQuant G-50 Micro Columns | Cytiva Biosciences | 28903408 | refered to as gel filtration-based purification kit |
| QIAquick gel extraction kit | Qiagen | 28506 | refered to as mini column-based gel-purification kit |
| RNeasy MinElute cleanup kit | Qiagen | 74204 | refered to as mini column-based RNA purification kit |
| TSA Biotin kit | Akoya Biosciences (Perkin Elmer) | SAT700001EA | n/a |
| Oligonucleotides | |||
| 5’ – AAACTTCATCCTTACAGAATGG CAG – 3’ |
Integrated DNA Technologies | n/a | Olfr692 |
| 5’ – ACTGGCTTTGGGACAGTGTGAC – 3’ | Integrated DNA Technologies | n/a | |
| 5’- GGTAATATCTCCATTATCCTAGTT TCCC – 3’ |
Integrated DNA Technologies | n/a | Olfr124 |
| 5’ – TTGACCCAAAACTCCTTTGTTAG TG – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – ATGGGAGCTCTAAATCAAACAA GAG – 3’ |
Integrated DNA Technologies | n/a | Olfr1509 |
| 5’ – TAGAAAACCGATACCACCTTGTC G – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – TACATCCTGACTCAGCTGGGGA ACG – 3’ |
Integrated DNA Technologies | n/a | Olfr1512 |
| 5’ – GGGCACATAGTACACAGTAACA ATAGTC – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – GAGGAAGCTCACTTTTGGTTTG G – 3’ |
Integrated DNA Technologies | n/a | Olfr78 |
| 5’ – CAGCTTCAATGTCCTTGTCACA G – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – TGGGTTGGAGGCTTATCATACC TG – 3’ |
Integrated DNA Technologies | n/a | Olfr691 |
| 5’ – AAGAACAACACAGAGTCTTGAT GTC – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – AGAAGTAACTAACACCACTCAT GGC – 3’ |
Integrated DNA Technologies | n/a | Olfr638 |
| 5’ – TTAGTGCACCTTTCTTTGCAAC – 3’ | Integrated DNA Technologies | n/a | |
| 5’ – TAACAGCTCTTCCCATCCCCTG TTC – 3’ |
Integrated DNA Technologies | n/a | Olfr569 |
| 5’ – TAGGGTTGAGCATGGGAGGAAC AAGC – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – CACTGGATCAACTCTAGCAGCA CTG – 3’ |
Integrated DNA Technologies | n/a | Vmn2r1 |
| 5’ – CTGCCCTTCTTGACATCTGCTG AG – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – ATCGGATCCACTGCTTTAGCATT TCTTACAGGACAG – 3’ |
Integrated DNA Technologies | n/a | Vmn2r2 |
| 5’ – ATCCTCGAGTCATGCCTCTCCAT AAGCAAGGAATTCCAC – 3' |
Integrated DNA Technologies | n/a | |
| 5’ – TAGGAAGCTATTTGCCTTGTTTC CAC – 3’ |
Integrated DNA Technologies | n/a | Vmn2r13 |
| 5’ – AGGAGATTTTACCAACCAGATTC CAG – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – CTCTAAGAACAGCAGTAAAATGG ATCT – 3’ |
Integrated DNA Technologies | n/a | Vmn2r89 |
| 5’ – ATGGGAATGACCAACTTAGGTGC A – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – ATCCCATGGCTGAGAACATGTGC TTCTGGAG – 3’ |
Integrated DNA Technologies | n/a | Vmn2r118 |
| 5’ – ATCCTCGAGTCAGTCTGCATAAG CCAGATATGTCAC – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – ATCGGATCCGCTGATTTTATTTCT CCCAGATGCTTTTGG – 3’ |
Integrated DNA Technologies | n/a | Vmn2r116 |
| 5’ – ATCCTCGAGTCATGGTTCTTCAT AGCTGAGAAATACAAC – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – TGGGTGTCTTCTTTCTCCTCAA GA – 3’ |
Integrated DNA Technologies | n/a | Vmn2r28 |
| 5’ – GGTGACCCATATTCTCTGTATAA CTGT – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – GATGTTCATTTTCATGAGAGTCT TCC – 3’ |
Integrated DNA Technologies | n/a | Vmn2r41 |
| 5’ – CATTTGTGGATGACATCACAATT TGG – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – TTTATGGCAAATTTCACTGATCCC G – 3’ |
Integrated DNA Technologies | n/a | Vmn2r46 |
| 5’ – AGTGGGTCTTTCTTAGAAAGGAG TG – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – ACATGAACCAGAATTTGAAGCAG GC – 3’ |
Integrated DNA Technologies | n/a | Vmn2r69 |
| 5’ – GCCAAGAAAGCTACAGTGAAAC C – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – AGGTGAAGAAATGGTATTCTTCC AG – 3’ |
Integrated DNA Technologies | n/a | Vmn2r58 |
| ACTGTGGCCTTGAATGCAATAACT – 3’ | Integrated DNA Technologies | n/a | |
| 5’ – TTCCTAAAGAACACCCTACTGA AGCATCG – 3’ |
Integrated DNA Technologies | n/a | Vmn2r90 |
| 5’ – CATATTCCACAGAAGAGAAGT TGGAC – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – TTGAGGTGAGAGTCAACAGT TTAGAC – 3’ |
Integrated DNA Technologies | n/a | Vmn2r107 |
| 5’ – CCCTTGTTGCACAAAATGAT GATGTGA – 3’ |
Integrated DNA Technologies | n/a | |
| 5’ – ATCCCATGGAGTCAGAGTAT CTACTACACCATGATGG – 3’ |
Integrated DNA Technologies | n/a | Vmn2r83 |
| 5’ – ATCCTCGAGTCAATCATTAT AGTCCAGAAAGGTGACAG – 3’ |
Integrated DNA Technologies | n/a | |
| Recombinant DNA | |||
| pGEM-T-Easy vector | Promega | A1360 | reccommended PCR cloning vector |
| Other materials | |||
| 1mL syringes | Fisher Scientific | 14-829-10F | n/a |
| Conical tubes (15 mL and 50 mL) | Fisher Scientific | 14-432-22, 14-959-49B | n/a |
| Coplin jars | Fisher Scientific | 12-567-099, 07-200-81 | n/a |
| Cryostat | Leica Biosystems | CMS 1850 | n/a |
| Dissecting tools and forceps | Roboz | RS-6802, RS-8124, RS-7110, RS-5111 | n/a |
| Dry bath | n/a | n/a | n/a |
| Electrophoresis equipment | Fisher Scientific | 09-528-110B | n/a |
| Fine point paintbrushes | Winsor & Newton | 10269097 | n/a |
| Fluorescence or confocal microscope | Leica Microsystems | TCS SP5II | n/a |
| Heated plate | Fisher Scientific | HP88850200 | n/a |
| Humidified chamber (if used at higher temperatures, it will need to be sealed inside a plastic Tupperware container) | Thermo Fisher Scientific | 22-045-034 | n/a |
| Lint-free laboratory Kimwipes | Kimberly-Clark | 34120 | refered to as lint-free laboratory tissue paper |
| Microcentrifuge | Eppendorf | 5401000013 | n/a |
| Mouse cages | InnoVive | M-BTM, MVX1 | n/a |
| PCR Thermocycler | Thermo Fisher Scientific | 4375786 | n/a |
| Pipette p1000, p200, and p20 disposable tips | Fisher Scientific | 02-707-408, 02-707-411, 02-707-438 | n/a |
| Plastic histology embedding mold | Thermo Fisher Scientific | 22-19 | n/a |
| Qubit 4 Fluorometer | Thermo Fisher Scientific | Q33238 | refered to as highly sensitive fluorometric method |
| Razor blades or scalpels | Fisher Scientific | 12-640 | n/a |
| RNA tapestation or BioAnalyzer | Agilent | 4200 TapeStation System or 2100 Bioanalyzer Instrument | n/a |
| RNase-free glass or plastic graduated cylinders and beakers | Fisher Scientific | 10-462-833, 02-555-25B, 02-555-25D | n/a |
| Stereomicroscope | Leica Microsystems | M80 | n/a |
| SuperFrost Plus microscope slides | Thermo Fisher Scientific | 12-550-15 | referred to as positively charged microscope slides |
| Parafilm | Bemis Company | PM999 | refered to as thermoplastic laboratory film |
| Water bath | Thermo Fisher Scientific | TSCIR19 | n/a |
Referências
- Stowers, L., Holy, T. E., Meister, M., Dulac, C., Koentges, G. Loss of sex discrimination and male-male aggression in mice deficient for TRP2. Science. 295 (5559), 1493-1500 (2002).
- Chamero, P., et al. Identification of protein pheromones that promote aggressive behaviour. Nature. 450 (7171), 899-902 (2007).
- Papes, F., Logan, D. W., Stowers, L. The vomeronasal organ mediates interspecies defensive behaviors through detection of protein pheromone homologs. Cell. 141 (4), 692-703 (2010).
- Munger, S. D., Leinders-Zufall, T., Zufall, F. Subsystem organization of the mammalian sense of smell. Annual review of physiology. 71, 115-140 (2009).
- Chess, A., et al. Allelic inactivation regulates olfactory receptor gene expression. Cell. 78, 823-834 (1994).
- Mombaerts, P. Genes and ligands for odorant, vomeronasal and taste receptors. Nature Reviews Neuroscience. 5, 263-278 (2004).
- Malnic, B., Hirono, J., Sato, T., Buck, L. B. Combinatorial receptor codes for odors. Cell. 96 (5), 713-723 (1999).
- Dulac, C., Axel, R. A novel family of genes encoding putative pheromone receptors in mammals. Cell. 83 (2), 195-206 (1995).
- Herrada, G., Dulac, C. A novel family of putative pheromone receptors in mammals with a topographically organized and sexually dimorphic distribution. Cell. 90 (4), 763-773 (1997).
- Martini, S., Silvotti, L., Shirazi, A., Ryba, N. J. P., Tirindelli, R. Co-Expression of Putative Pheromone Receptors in the Sensory Neurons of the Vomeronasal Organ. J. Neurosci. 21 (3), 843-848 (2001).
- Silvotti, L., Moiani, A., Gatti, R., Tirindelli, R. Combinatorial co-expression of pheromone receptors, V2Rs. Journal of Neurochemistry. 103 (5), 1753-1763 (2007).
- Ishii, T., Mombaerts, P. Expression of nonclassical class I major histocompatibility genes defines a tripartite organization of the mouse vomeronasal system. Journal of Neuroscience. 28 (10), 2332-2341 (2008).
- Saraiva, L. R., et al. Hierarchical deconstruction of mouse olfactory sensory neurons: From whole mucosa to single-cell RNA-seq. Scientific Reports. 5, (2015).
- Hanchate, N. K., et al. Single-cell transcriptomics reveals receptor transformations during olfactory neurogenesis. Science. 350, 1251-1255 (2015).
- Matsunami, H. Functional expression of mammalian odorant receptors. Chemical Senses. 30, 95-96 (2005).
- Isogai, Y., et al. Molecular organization of vomeronasal chemoreception. Nature. 478 (7368), 241-245 (2011).
- Xu, L., Li, W., Voleti, V., Zou, D. J., Hillman, E. M. C., Firestein, S. Widespread receptor-driven modulation in peripheral olfactory coding. Science. 368 (6487), (2020).
- Pfister, P., et al. Article Odorant Receptor Inhibition Is Fundamental to Odor Encoding Odorant Receptor Inhibition Is Fundamental to Odor Encoding. Current Biology. , 1-14 (2020).
- de March, C. A., Titlow, W. B., Sengoku, T., Breheny, P., Matsunami, H., McClintock, T. S. Modulation of the combinatorial code of odorant receptor response patterns in odorant mixtures. Molecular and Cellular Neuroscience. 104, 103469 (2020).
- Nakahara, T. S., Carvalho, V. M. A., Souza, M. A. A., Trintinalia, G. Z., Papes, F. Detection of Activated Mouse Neurons with Temporal Resolution via Dual c-Fos Staining. STAR Protocols. 1 (3), 100153 (2020).
- Nakahara, T. S., et al. Detection of pup odors by non-canonical adult vomeronasal neurons expressing an odorant receptor gene is influenced by sex and parenting status. BMC biology. 14 (1), 12 (2016).
- Carvalho, V. M. A., et al. Lack of spatial segregation in the representation of pheromones and kairomones in the mouse medial amygdala. Frontiers in Neuroscience. 9, 1-19 (2015).
- Carvalho, V. M. A., et al. Representation of Olfactory Information in Organized Active Neural Ensembles in the Hypothalamus. Cell Reports. 32 (8), 108061 (2020).
- Papes, F., Nakahara, T. S., Camargo, A. P. Behavioral assays in the study of olfaction: a practical guide. Methods in Molecular Biology, v1820: Olfactory receptors. , 289-388 (2018).
- Jiang, Y., Gong, N. N., Hu, X. S., Ni, M. J., Pasi, R., Matsunami, H. Molecular profiling of activated olfactory neurons identifies odorant receptors responding to odors in vivo. Nature Neuroscience. 18, 1446-1454 (2015).
- Von Der Weid, B., et al. Large-scale transcriptional profiling of chemosensory neurons identifies receptor-ligand pairs in vivo. Neurociência. 18 (10), (2015).

.