Site Specific Lysine Acetylation of Histones for Nucleosome Reconstitution using Genetic Code Expansion in Escherichia coli
Summary
Here we present a method to express acetylated histone proteins using genetic code expansion and assemble reconstituted nucleosomes in vitro.
Abstract
Acetylated histone proteins can be easily expressed in Escherichia coli encoding a mutant, Nε-acetyl-lysine (AcK)-specific Methanosarcina mazi pyrrolysine tRNA-synthetase (MmAcKRS1) and its cognate tRNA (tRNAPyl) to assemble reconstituted mononucleosomes with site specific acetylated histones. MmAcKRS1 and tRNAPyl deliver AcK at an amber mutation site in the mRNA of choice during translation in Escherichia coli. This technique has been used extensively to incorporate AcK at H3 lysine sites. Pyrrolysyl-tRNA synthetase (PylRS) can also be easily evolved to incorporate other noncanonical amino acids (ncAAs) for site specific protein modification or functionalization. Here we detail a method to incorporate AcK using the MmAcKRS1 system into histone H3 and integrate acetylated H3 proteins into reconstituted mononucleosomes. Acetylated reconstituted mononucleosomes can be used in biochemical and binding assays, structure determination, and more. Obtaining modified mononucleosomes is crucial for designing experiments related to discovering new interactions and understanding epigenetics.
Introduction
We have utilized PylRS and tRNAPyl to synthesize and assemble reconstituted mononucleosomes with site specific acetylated histones. PylRS has proven invaluable as a genetic code expansion tool to produce proteins with post translational modifications (PTMs) and has been genetically evolved to incorporate about 200 different ncAAs. PylRS incorporates at an amber stop codon, removing competition from other amino acids during translation. PylRS was first discovered in methanogenic archaea, and has since been utilized in chemical biology to incorporate novel reactive chemical groups into proteins1,2.
MmAcKRS1 was evolved from Methanosarcina mazei PylRS and frequently used in our laboratory for the synthesis of acetylated proteins3,4,5,6. MmAcKRS1 delivers AcK at an amber mutation site in the mRNA of choice during translation in Escherichia coli. This technique has been previously used to incorporate AcK at K4, K9, K14, K18, K23, K27, K36, K56, K64, and K79 histone H3 lysine sites to study the activity of Sirtuin 1, 2, 6, and 7 on acetylated mononucleosomes4,5,6. Here we detail this method to express acetylated histones and reconstitute acetylated nucleosomes.
Protocol
1. Plasmid construction
- Begin by deciding which histone protein will be acetylated and at which lysine site. Mutate the site to the amber stop codon (TAG) using site directed mutagenesis.
NOTE: There are four previously designed plasmids utilized for expression of histone proteins. All four histone proteins were cloned into the pETDuet-1 vector with an N-terminal histidine tag. Histone H4 also includes a SUMO tag, the origin of replication colE1 with a copy number of approximately 40, ampicillin resistant, and a T7 promoter.- Design forward and reverse primers that contain a TAG mutation at the desired site (replacing an existing lysine codon with the amber stop codon) in one of the four histone protein plasmids.
- Use whole-plasmid PCR to amplify the TAG containing plasmid. Determine an appropriate annealing temperature for the primers. In general, the melting temperature (Tm) of the primers minus 5 °C will be sufficient. If difficulty is experienced obtaining a PCR product, a temperature gradient around Tm – 5 °C can be used to optimize the annealing temperature for amplification.
NOTE: In this experiment an in-house expressed PFU polymerase was used for 30 cycles with the following conditions: 94 °C for 30 s (denaturation), annealing temperature for 30 s, and 72 °C for 6 min (or 1 min per kilobase-pairs). For more details on this type of PCR method see Liu and Naismith7. - Clean up the PCR product with a PCR clean up kit by following the manufacturer's protocol. Analyze the PCR product by agarose gel electrophoresis and subsequent ethidium bromide staining.
- Ligate the plasmid by incubating with T4 ligase overnight at 16 °C.
- First transform the TAG containing plasmid (AmpR) into chemically competent Escherichia coli. Pick a minimum of 4 colonies from the transformation and purify the plasmid. Make cell stocks of each with glycerol using standard methods and store at -80 °C. Send for sequencing to confirm the desired TAG mutation.
- Once the sequence is confirmed, co-transform the TAG-containing plasmid (AmpR) with pEVOL-AckRS (ChlR) into chemically competent CobB (the only histone lysine deacetylase known in E. coli.) deletion BL21 cells using the heat shock method. pEVOL is a plasmid with a mid-copy-number to low-copy-number p15A origin.
NOTE: The pEVOL series of plasmids were constructed based on previous studies that showed an increased expression of the orthogonal aaRS/tRNATyr pair was effective in decreasing protein truncation and in increasing overall yields of mutant proteins8. If CobB deletion cells are not accessible, proceed with chemically competent BL21 cells. CobB is a Sirtuin-like histone lysine deacetylase and Nicotinamide can be added at a 5 mM final concentration during histone protein expression to inhibit CobB as an alternative approach9. - Make a cell stock and store at -80 °C.
2. Acetylated histone protein expression
- Prepare 1 L of autoclaved 2YT media (or volume of choice) in a culture flask.
- Inoculate 20 mL of 2YT media containing the appropriate antibiotics with the co-transformed cell stock that has the TAG-containing plasmid (AmpR) and pEVOL-AckRS (ChlR) and grow at 37 °C to OD = 0.6 (4-6 h).
- Add the appropriate antibiotics to the 1 L of autoclaved 2YT media and inoculate with the 20 mL starter culture. Grow at 37 °C to OD = 0.6-0.8 (2-3 h).
- Add inducing agents and acetyllysine (AcK) to the final concentrations of IPTG 1 mM, 0.2% arabinose, and AcK 5 mM.
- Grow the culture at 37 °C for 6-8 h.
- Pellet cells at 2,700 x g for 15 min.
- Store the cell pellet at -80 °C overnight.
- From this step onwards keep the sample on ice all times. Dissolve the cell pellet in 50 mL of histone lysis buffer per 1 L of culture.
- Sonicate according to the following cycle: 1 s on, 1 s off, total on time: 3 min at 60% amplitude.
- Pellet inclusion bodies at 41,600 x g for 45 min.
- Discard the supernatant and resuspend inclusion bodies in 30 mL of histone lysis buffer. Pellet inclusion bodes at 41,600 x g for 30 min.
- Discard the supernatant and resuspend inclusion bodies in 30 mL of pellet wash buffer. Pellet inclusion bodies at 41,600 x g for 30 min.
- Discard the supernatant and dissolve inclusion bodies in 25 mL of 6 M Guanidine Hydrochloride (GuHCl) buffer. Incubate with agitation at 37 °C for 1 h.
NOTE: If needed, inclusion bodies can be incubated with agitation overnight at 4 °C. - Pellet inclusion bodes at 41,600 x g for 45 min. Incubate the supernatant with 1 mL of Ni-NTA resin equilibrated with 6 M GuHCl buffer for 2 h.
- Proceed with Ni-NTA purification under denatured conditions. Wash the column with 3 column volumes of column wash buffer. Elute acetylated histone protein with 10 mL of elution buffer.
NOTE: Attempting to concentrate the protein here is an option but acetylated histones are very unpredictable and can easily precipitate. It is not recommended to concentrate past 2 mL in total volume. - Extensively dialyze the acetylated histone protein against 5% acetic acid buffer to remove salts. Dialyze for 3 h at a time at 4 °C, exchanging for fresh buffer a minimum of 6 times.
NOTE: For improved protein purity, there is an option to dialyze against pure water. However, this causes significant precipitation and reduction in yield which may be undesirable for low-yielding acetylated histone proteins. - Aliquot and lyophilize protein, and store indefinitely at -80 °C. Wild type histone proteins generally yield 10-50 mg/L whereas acetylated histone proteins yield less than 10 mg/L depending on the specific lysine site. For example, H3K79Ac averages 5 mg/L.
3. Wild-type histone protein expression
- To assemble full nucleosomes, express all 4 histone proteins. When expressing and purifying wild type histones the protocol is the same as the acetylated histones except the following:
- Do not perform co-transformation. Do not use pEVOL-AcKRS for wild type histone expression. Adjust antibiotics accordingly.
- Do not use a CobB deletion cell line or add Nicotinamide, AcK, or arabinose during induction of cellular expression.
4. Preparation of 601 DNA
NOTE: A previously designed optimized DNA sequence to direct nucleosome positioning is assembled with histone octamers to produce mononucleosome with high efficiency. This sequence is referred to as 601 DNA or the Widom sequence. This sequence has become the standard DNA sequence for in vitro studies of nucleosomes from chromatin remodeling assays to single molecule measurements10.
- To prepare significant quantities of 601 DNA for nucleosome assembly, transform pGEM-3z/601 into Top 10 cells and grow 10 mL of culture for plasmid amplification and extraction. Use a plasmid extraction kit and follow manufacturer's recommendation.
- Use an in-house expressed PFU polymerase (more efficient) for this amplification protocol. Set up a PCR reaction: For 250 µL reaction, use 207.5 µL autoclaved MQ H2O, 25 µL 10x PFU Buffer, 5 µL Forward Primer: ctggagaatcccggtgccg, 5 µL Reverse Primer: acaggatgtatatatctgacacg, 5 µL dNTP mix, 2.5 µL PFU enzyme. We typically run 60 reactions at once to get enough DNA to assemble.
- Use an annealing temperature of 52 °C for 30 s and an extension time of 30 s if using PFU polymerase. Otherwise follow the manufacturers recommended conditions.
- Once PCR is done, use a PCR clean up kit of choice to purify the PCR product.
5. Assembly of histone octamer
- Dissolve aliquots of histone protein pellets H2A, H2B, H3, and H4 so that there is a separate stock of each histone protein in GuHCl Buffer with a total volume of 100 µL.
- Calculate the concentration of each histone protein by measuring the A280 absorbance.
- If absorbance is greater than 1 for any protein, dilute with GuHCl buffer to get a more accurate concentration.
- Combine H2A and H2B proteins in a 1:1 molar ratio and dilute to a total protein concentration of 4 µg/µL. Repeat for H3 and H4.
- Dialyze sequentially at 4 °C against 2 M TE buffer overnight, 1 M TE buffer for 2 h and 0.5 M TE buffer for 5 h.
NOTE: The second and third steps of dialysis causes unstable conformations of protein to precipitate out. It is expected to see heavy precipitation at these steps. - Remove precipitates by centrifugation at 16,800 x g at 4 °C.
- Again, calculate the concentration of histone dimers (H2A/H2B mixture) and tetramers (H3/H4 mixture) measuring the A280 absorbance.
- Mix dimers and tetramers in a 1:1 molar ratio and adjust NaCl to 2 M by the addition of solid NaCl.
- Histone octamer can be stored at 4 °C and is more stable than dimers and tetramers. Never freeze histone octamer as this can cause disassembly.
- If assembling with acetylated histones, simply replace the wild type protein with the acetylated protein in the procedure.
6. Nucleosome assembly
- Use 100x TE buffer and solid NaCl to adjust 601 DNA to 2M TE buffer. Add the 601 DNA in 2M TE buffer to histone octamer in a molar ratio of 0.85:1 to 0.90:1. A lower ratio of DNA may be added if free DNA is present in the gel shift analysis.
- Transfer the DNA-histone mixture a dialysis bag and place in about 200 mL of 2M TE buffer (or more if assembling multiple samples) and very gently stir at 4 °C.
- Set a peristaltic pump to purge to slowly drip in no salt TE buffer. When the volume has roughly doubled, pour out half the volume. Do this at least 4 times in total. With a pump this can take 4-8 h depending on the starting volume. Nucleosomes form when the salt concentration is reduced to 150 mM (measured by salinity meter).
- After the salt concentration is reduced to 150 mM, dialyze against a 20 mM TE buffer overnight.
- Remove precipitates by centrifugation and measure the concentration of the nucleosomes by A260 reading using a plate reader.
- Add His-TEV protease (TEV: nucleosome 1:30, w:w ) and incubate overnight at 4 °C to remove all histidine tags. Remove the histidine tag impurities from the nucleosome solution by Ni-NTA resin pull down.
- To position the nucleosome and homogenize the sample, incubate at 60 °C for 1 h.
NOTE: For acetylated sites that are particularly unstable, this step may not be advisable. - Store nucleosomes for short term (a few weeks) at 4 °C.
- For long-term storage, dialyze nucleosomes against storage buffer and store at -80 °C.
Representative Results
Dimers, tetramers, and octamers can be assessed by running a 12% SDS PAGE gel (Figure 1 and Figure 2). Here you can see that some of the acetylated tetramers have a lower yield than others (Figure 1). In fact, the closer to the core region, the lower the yield observed. This is most likely due to the acetylation interfering with the assembling of the tetramer the closer you get to the core regions. A similar affect is observed after assembling octamers (Figure 2). After assembling octamers with 601 DNA sequence, the nucleosome can be assessed using a 5% 1x TBE Native PAGE gel followed with staining with ethidium bromide (Figure 3). Before TEV digestion the nucleosome band is observed near the 10k base pair mark. After TEV digestions the nucleosome band shifts down relative to the ladder. Before thermal positioning, the observed nucleosome bands will be very broad, and possibly streak (Figure 4).

Figure 1: Histone dimers and tetramers. Representative 12% SDS PAGE gel of wild type histone dimer (lane 2) and H3 acetylated tetramers (lanes 3-9). Please click here to view a larger version of this figure.

Figure 2: Histone octamers. Representative 12% SDS PAGE gel of H3 acetylated histone octamers (lanes 2-11). Please click here to view a larger version of this figure.
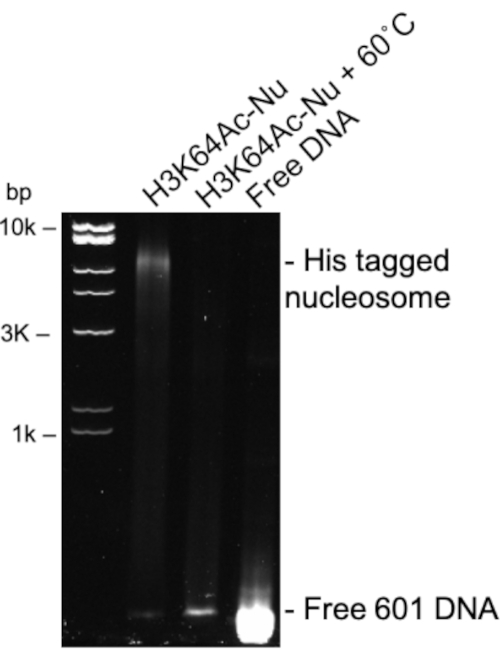
Figure 3: Assembled his tagged H3K64Ac nucleosome complex. Representative 1x TBE Native PAGE gel of H3K64Ac nucleosome complex (lane 2) compared to free 601 DNA (Lane 4) visualized by ethidium bromide. Lane 3 shows H3K64Ac nucleosome after incubation at 60 ˚C for one hour. It is observed in lane 3 that all nucleosome has disassembled. Please click here to view a larger version of this figure.

Figure 4: Assembled TEV digested nucleosome complex. Representative 1x TBE Native PAGE gel of wild type TEV digested nucleosome complex, and free 601 DNA. A comparison of before and after nucleosome thermal positioning. Please click here to view a larger version of this figure.
Discussion
It is essential to follow this protocol in every detail during an experiment. Nucleosomes are not very stable and much trial and error has gone into determining this protocol. It is key to remove precipitates at every step (or whenever observed) because particulates can easily interfere with the assembling processes. Always keep histone samples on ice. If nucleosomes are stored at 4 °C for too long, they can spontaneously disassemble. Be sure to check any samples by Native PAGE if stored at 4 °C for more than 2 weeks before use in any experiment. Avoid vortexing octamers and nucleosomes as this can also cause them to disassemble. If issues are encountered assembling dimers, tetramers, or octamers this is usually indicative of poor protein quality. Check the protein quality by 15% SDS-PAGE. One option if protein purity is low is to dialyze the protein against pure water. This will cause heavy precipitation and greatly reduce protein yield, but will produce a purer sample. The incorporation and nucleosomal stability of each histone acetylation site varies widely. In general, the closer the site is to the N-terminal domain, the lower the yield during histone protein expression. For nucleosome stability, the closer the acetylation site is to the nucleosome core or DNA binding sites, the less stable the assembled nucleosome. If stability issues are encountered, it is crucial to always keep the nucleosome sample on ice. It may be necessary to omit the nucleosome positioning step at 60 °C as this may cause disassembly or precipitation. A major limitation of acetylated nucleosomes is their instability. It may be impossible to preform assays at higher temperatures such as 37 °C without causing nucleosomal precipitation. If experimental procedures allow, perform them at 4 °C to prevent precipitation of the acetylated nucleosome.
This method of producing nucleosomes is particularly useful for producing modified nucleosomes for binding experiments and structure determinations. This method produces nucleosomes with higher purity and a known nucleosomal DNA sequence, which eliminates confounding variables resulting in better controlled experiments and higher resolution images. It is crucial for structure determinations that the nucleosome sample is homogenous with a known DNA sequence, otherwise it will be all but impossible to obtain high resolution images.
There is an abundance of potential applications of this method. Specifically, in the field of epigenetics. It can be incredibly difficult to obtain modified proteins. Obtaining modified proteins is crucial for probing the structure and function of the writers, readers, and erasers in the many epigenetic pathways that are poorly understood. We have previously utilized this technique to probe the accessibility of nucleosomal DNA to Pst1 digestion at the H3 acetylation sites K18, K36, and K56 by assembling each mutant octamer with a modified 601 DNA sequence that contains a Pst1 digestion site. This technique can be further modified to incorporate NCAAs other than AcK. PylRS can be engineered to incorporate a host of other NCAAs. We have also incorporated Nε-(7-azidoheptanoyl)-L-lysine (AzHeK) by amber codon in Escherichia coli for recombinant expression of several AzHeK-containing histone H3 proteins to investigate the histone deacylation activity of SIRT7 at several acetylation sites. This approach revealed that SIRT7 has high activity towards deacylation of H3K36 and is also catalytically active to deacylate H3K37. H3K36 deacylation was further showed to be nucleosome dependent and can be significantly enhanced by adding a short double-stranded DNA to the acyl-nucleosome substrate that mimics the bridging DNA between nucleosomes in native chromatin6.
This method can also be modified to produce wild type nucleosomes with no modifications which can be useful in a variety of scenarios. Our group recently published a structure in collaboration with Dr. Pingwei Li's group showing the tight binding of cyclic GMP-AMP synthase (cGAS) to a negatively charged acidic patch formed by histone H2A and H2B via its second DNA binding site11. cGAS is a dsDNA sensor that catalyzes the synthesis of a cyclic dinucleotide cGAMP, which mediates the induction of type I interferons through the STING-TBK1-IRF3 signaling axis12,13,14,15,16.
The benefit of using fully assembled nucleosomes is it more closely resembles the native state of nucleosomes serving as a better model to probe the activities or binding abilities of any number of epigenetic-related proteins. There are numerous examples of limited or entirely absent enzyme activity towards naked DNA or histone peptide substrates that when probed with nucleosome substrates drastically changes results. As with the example mentioned above, a novel deacylation site was discovered for SIRT7 by using a nucleosome substrate that would have otherwise been unknown. It is crucial to use native substrates when probing these types of systems. This technique can be used to probe the reading, writing, and erasing activity or binding capability of any modification that can be incorporated by the pyrrolysine genetic code expansion technique. The pyrrolysyl-tRNA synthetase is already natively promiscuous and has been engineered for a host of other substrates already opening the door for using this technique in any number of systems making the primary limiting factor the creativity of the researcher.
Declarações
The authors have nothing to disclose.
Acknowledgements
We would like to thank Dr. Wesley Wang for laying the groundwork for this protocol and his valuable mentorship. This work was partially supported by National Institutes of Health (Grants R01GM127575 and R01GM121584) and Welch Foundation (Grant A-1715).
Materials
| 0.5 M TE Buffer | NA | NA | 0.5 M NaCl, 20 mM Tris, 1 mM EDTA, pH 7.8 |
| 1 M TE Buffer | NA | NA | 1 M NaCl, 20 mM Tris, 1 mM EDTA, pH 7.8 |
| 100x TE Buffer | NA | NA | |
| 2 M TE Buffer | NA | NA | 2 M NaCl, 20 mM Tris, 1 mM EDTA, pH 7.8 |
| 20 mM TE Buffer | NA | NA | 20 mM NaCl, 20 mM Tris, 1 mM EDTA, pH 7.8 |
| 6 M GuHCl | 6M guanidinium chloride, 20 mM Tris, 500 mM NaCl, pH 8.0 | ||
| Acetyllysine | |||
| Column Wash Buffer | NA | NA | 6 M urea, 500 mM NaCl, 20 mM Tris, 20 mM imidazole pH 7.8 |
| Elution Buffer | NA | NA | |
| Fisherbrand Variable-Flow Chemical Transfer Pump | Fischer Scientific | 15-077-67 | |
| His-TEV protease | |||
| Histone Lysis Buffer | NA | NA | 60 mM Tris, 100 mM NaCl, 0.5% Triton-X100 (v/v), 1 mM PMSF pH 8.0 |
| Ni-NTA Resin | 6 M urea, 500 mM NaCl, 20 mM Tris, 250 mM imidazole, pH 7.8 | ||
| PCR Clean-Up Kit | Epoch Life Sicences | 2360050 | |
| Pellet Wash Buffer | NA | NA | 60 mM Tris, 100 mM NaCl, pH 8.0 |
| petDUET-His-SUMO-TEV-H4 | |||
| petDUET-His-TEV-H2A | |||
| petDUET-His-TEV-H2B | |||
| petDUET-His-TEV-H3 | |||
| pEVOL-AckRS | Addgene | 137976 | |
| pGEM-3z/601 | Addgene | 26656 | |
| Storage Buffer | NA | NA | 20 mM NaCl, 20 mM Tris, 20 mM NaCl, 1 mM EDTA, 20% glycerol, pH 7.8 |
| Thermocycler |
Referências
- Srinivasan, G., James, C. M., Krzycki, J. A. Pyrrolysine encoded by UAG in Archaea: Charging of a UAG-Decoding specialized tRNA. Science. 296 (5572), 1459-1462 (2002).
- Wan, W., Tharp, J. M., Liu, W. R. Pyrrolysyl-tRNA synthetase: an ordinary enzyme but an outstanding genetic code expansion tool. Biochimica Et Biophysica Acta. 1844 (6), 1059-1070 (2014).
- Umehara, T., et al. N-Acetyl lysyl-tRNA synthetases evolved by a CcdB-based selection possess N-acetyl lysine specificity in vitro and in vivo. FEBS Letters. 586 (6), 729-733 (2012).
- Hsu, W. W., Wu, B., Liu, W. R. Sirtuins 1 and 2 are universal histone deacetylases. ACS Chemical Biology. 11 (3), 792-799 (2016).
- Wang, W. W., Zeng, Y., Wu, B., Deiters, A., Liu, W. R. A chemical biology approach to reveal Sirt6-targeted histone H3 sites in nucleosomes. ACS Chemical Biology. 11 (7), 1973-1981 (2016).
- Wang, W. W., et al. A click chemistry approach reveals the chromatin-dependent histone H3K36 deacylase nature of SIRT7. Journal of the American Chemical Society. 141 (6), 2462-2473 (2019).
- Liu, H., Naismith, J. H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnology. 8 (1), 91 (2008).
- Young, T. S., Ahmad, I., Yin, J. A., Schultz, P. G. An enhanced system for unnatural amino acid mutagenesis in E. coli. Journal of Molecular Biology. 395 (2), 361-374 (2010).
- Gallego-Jara, J., Écija Conesa, A., de Diego Puente, T., Lozano Terol, G., Cánovas Díaz, M. Characterization of CobB kinetics and inhibition by nicotinamide. PLoS One. 12 (12), 0189689 (2017).
- Lowary, P. T., Widom, J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. Journal of Molecular Biology. 276 (1), 19-42 (1998).
- Zhao, B., et al. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature. 587 (7835), 673-677 (2020).
- Burdette, D. L., Vance, R. E. STING and the innate immune response to nucleic acids in the cytosol. Nature Immunology. 14 (1), 19-26 (2013).
- Barber, G. N. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Current Opinion in Immunology. 23 (1), 10-20 (2011).
- Ablasser, A., et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 498 (7454), 380-384 (2013).
- Gao, P., et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 153 (5), 1094-1107 (2013).
- Zhao, B., et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature. 569 (7758), 718-722 (2019).
