Production of most eukaryotic messenger RNAs (mRNAs) involves removal of introns and ligation of exons in a nuclear process termed pre-mRNA splicing1. Two classes of RNA-protein complexes (RNPs) direct the processing of pre-messenger RNA into mature mRNA via spliceosomal complexes. One class, nascent pre-messenger RNPs, is formed co-transcriptionally by the binding of heterogeneous nuclear RNP proteins and other RNA-binding proteins, including some members of the SR family, yielding hnRNP complexes2. The second class, uracil-rich small nuclear RNPs (U snRNPs with U1, U2, U4, U5, and U6 snRNAs) is associated with U-specific and core proteins3,4. The U snRNPs interact in an ordered fashion with specific regions of pre-messenger RNPs in a dynamic remodeling pathway as introns are excised and exons are ligated to produce mature mRNPs5. Many additional nuclear proteins participate in these processing events6.
Galectin-1 (Gal1) and galectin-3 (Gal3) are two proteins that are required factors in the splicing pathway as shown by depletion-reconstitution studies7,8. Removal of both galectins from splicing competent nuclear extracts (NE) abolishes spliceosome assembly and splicing activity at an early step. Addition of either galectin to such a doubly depleted NE restores both activities. Gal1 and Gal3 are components of active spliceosomes as evidenced by specific immunoprecipitation of pre-mRNA, splicing intermediates, and mature mRNA by antiserum specific for either Gal1 or Gal39. Importantly, Gal3 associates with endogenous U snRNA containing particles in the NE outside the splicing pathway as shown by precipitation of snRNPs by anti-Gal3 antisera10. Finally, silencing of Gal3 in HeLa cells alters splicing patterns of numerous genes11.
In NE pre-incubated to disassemble preformed spliceosomes12, snRNPs are found in multiple complexes sedimenting in glycerol gradients from 7S to greater than 60S. Although glycerol gradient fractionation is a common technique for the isolation of spliceosomal complexes and components (see references13,14,15 for example), we have extended this method by characterizing specific fractions using antibody immunoprecipitations. An snRNP sedimenting at 10S contains only U1 snRNA along with Gal3. Immunoprecipitation of the 10S fraction with antisera specific for Gal3 or U1 snRNP co-precipitates both U1 and Gal3 indicating some of the U1 snRNP monoparticles are bound to Gal310. As U1 snRNP is the first complex that binds to pre-mRNP in spliceosomal assembly1,5, this step represents a potential entry site for Gal3 into the splicing pathway. On this basis, we showed that 10S Gal3-U1 snRNP monoparticles bound to anti-Gal3 containing beads restored splicing activity to a U1 snRNP depleted NE, establishing this complex as one mechanism by which Gal3 is recruited into the spliceosomal pathway16. This contrasts with attempts to isolate spliceosomes at specific stages in the splicing reaction and cataloging the associated factors17,18. In such studies, the presence of certain factors at some time point is ascertained but not the mechanism by which they were loaded.
We had previously described in detail the preparation of NE, the splicing substrate, the assembly of the splicing reaction mixture, and the analysis of products in our documentation of the role of galectins in pre-mRNA splicing19. We now describe the experimental procedures for fractionation of nuclear extracts to obtain a fraction enriched in Gal3 – U1 snRNP complex and for immuno-selection of the latter complex to reconstitute splicing activity in a U1-depleted nuclear extract.
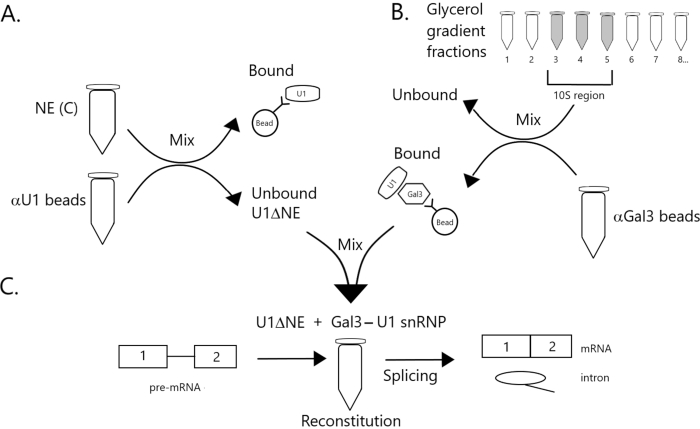
Figure 1: Schematic diagram illustrating the complementation of splicing activity in nuclear extract depleted of U1 snRNP by a Gal3-U1 snRNP complex on beads. (A) NE in Buffer C (NE(C)) is incubated with Protein A-Sepharose beads covalently coupled with anti-U1 snRNP (αU1 beads). The unbound fraction is depleted of U1 snRNP (U1ΔNE). (B) NE in Buffer D (NE(D)) is fractionated over a 12%-32% glycerol gradient by ultracentrifugation. Fractions corresponding to the 10S region (fractions 3-5) are combined and mixed with beads covalently coupled with anti-Gal3 antibodies (αGal3 beads). The material bound to the beads contains a Gal3-U1 snRNP monoparticle. (C) The Gal3-U1 snRNP complex from Part (B) is mixed with U1ΔNE from Part (A) in a splicing assay using 32P-labeled MINX pre-mRNA substrate and the intermediates and products of the splicing reaction are analyzed by gel electrophoresis and autoradiography. Please click here to view a larger version of this figure.
NE depleted of U1 snRNP (U1ΔNE from Section 2.2.6) and Gal3 – U1 snRNP complexes from the 10S region of the glycerol gradient immunoprecipitated by anti-Gal3 (step 3.2.7) were mixed in a splicing reaction. This reaction mixture contained U1 snRNA (Figure 2A, lane 3), as well as the U1-specific protein, U1-70K (Figure 2B, lane 3). As expected, the anti-Gal3 precipitated Gal3 (Figure 2B, lane 3). These components (U1 snRNA, U1-70K protein, and Gal3) were not found in the pre-immune (PI) control precipitation (Figure 2A, Figure 2B, lane 2). Compared to a non-depleted NE carried out as a positive control (Figure 2C, lane 1), U1ΔNE did not exhibit splicing activity (Figure 2C, lane 2). Splicing activity in the U1ΔNE could be reconstituted by the bead-bound Gal3 – U1 snRNP complex (Figure 2C, lane 6). Both products of the splicing reaction, ligated exons and excised intron lariat (highlighted by arrows on the right), as well as intermediates, exon 1 and lariat exon 2, were found. The components of fractions 3-5 were critical in restoring splicing to U1ΔNE. When fractions 3-5 were replaced by buffer alone (60% D) in the immunoprecipitation, no splicing activity could be observed on mixing with U1ΔNE (Figure 2C, lane 2), indicating that the anti-Gal3 beads were not responsible for the restoration of splicing activity in the latter. More persuasively, when fractions 3-5 were replaced by fraction 1 in the immunoprecipitation procedure and then added to U1ΔNE, no intermediates or products of the splicing reaction were found (Figure 2C, lane 4). Previous analysis had documented that the Gal3 in fraction 1 represented free Gal3 protein, not in association with any RNP complex10 and northern blotting of the material immunoprecipitated from fraction 1 failed to reveal any U1 snRNA16. Finally, immunoprecipitates from fraction1 and fractions 3-5 did not exhibit splicing activity when assayed in the absence of U1ΔNE (Figure 2C, lanes 3 and 5, respectively).
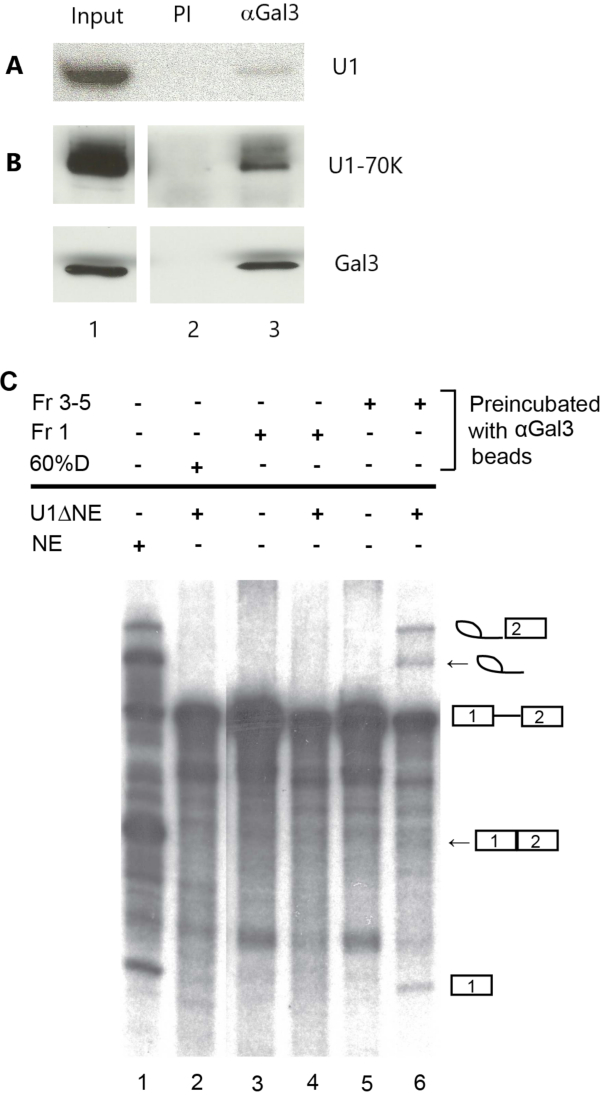
Figure 2: Representative results of the RNA and polypeptide compositions of the anti-Gal3 precipitate of the 10S region of the glycerol gradient and its ability to reconstitute splicing in nuclear extract depleted of U1 snRNP (U1ΔNE). (A) Northern blotting analysis of U1 snRNA bound to beads coupled with pre-immune serum (PI; lane 2) or to beads coupled with anti-Gal3 (αGal3; lane 3). Lane 1 represents 20% of the amount of combined glycerol gradient fractions subjected to immunoprecipitation. Lane 2 and lane 3 each represent 25% of the bound material eluted from the respective beads. (B) Western blotting analysis of polypeptides bound to beads coupled with pre-immune serum (lane 2) or anti-Gal3 (lane 3). Protein identities are indicated on the right. Lane 1 represents 20% of the amount of combined glycerol gradient fractions subjected to immunoprecipitation. Lane 2 and lane 3 each represent 75% of the bound material eluted from the respective beads. (C) Analysis of the ability to restore splicing activity to U1ΔNE by anti-Gal3 (αGal3) immunoprecipitates of glycerol gradient fraction 1 (Fr 1), fractions 3-5 (Fr 3-5), or 60% Buffer D (60% D). The + or – sign above the bolded solid line indicates the presence or absence of each of these precipitates in the splicing reaction mixture. The + or – sign below the bolded solid line indicates the presence or absence of U1ΔNE (or NE in Buffer D). Lane 1: 4 µL NE in a 12 µL splicing assay, 100% of which were subjected to gel analysis. For lanes 2-6, the total volume of the splicing assay was 24 µL, 50% of which were subjected to gel analysis. Lane 2: 8 µL U1ΔNE plus beads from precipitation of 60% D. Lane 3: beads from precipitation of Fr 1. Lane 4: 8 µL U1ΔNE plus beads from precipitation of Fr 1. Lane 5: beads from precipitation of Fr 3-5. Lane 6: 8 µL U1ΔNE plus beads from precipitation of Fr 3-5. The positions of migration of the pre-mRNA substrate, the intermediates, and the products (intron lariat and ligated exons, highlighted by arrows) are indicated on the right. The data in Panel C are derived from the same experiment as shown in Panel A, Figure 2 of Haudek et al.16. Please click here to view a larger version of this figure.
The results shown in Figure 2 were obtained with anti-Gal3 (#49). The same results could be observed with another anti-Gal3 (#24) but the pre-immune serum from rabbit #49 failed to reconstitute splicing in U1ΔNE following incubation with fractions 3-516. All of these results strongly suggest that the pre-mRNA substrate can bind to the 10S Gal3 – U1 snRNP particle immobilized on beads and that the ternary complex is functional in the splicing pathway.
