Previous work reported that the HeV-derived HeV1 (DFANTFLP) peptide was presented by Ptal-N*01:0110,19. Herein, the binding capacity of this peptide to Ptal-N*01:01 with homologous bat β2m (bβ2m) and heterologous human β2m (hβ2m) (Figure 1C,1D) was evaluated. Crystals with higher resolution were formed, respectively (Figure 1E,1F). A crystal is formed from the Ptal-N*01:01/HeV1 complex, which was formed through renaturation with bβ2m, and the resolution is 2.31 Å. A crystal is formed from the Ptal-N*01:01/HeV1 complex, which was formed through renaturation with hβ2m, and the resolution is 1.6 Å. The Ptal-N*01:01/HeV1 complex was successfully formed through renaturation with both bβ2m and hβ2m (Figure 1C,1D). In this context, we showed that the Ptal-N*01:01/HeV1 complex was not formed without the presence of β2m (Figure 1A) and the H-2Kd that fold correctly through the hβ2m (Figure 1B).
The structures of Ptal-N*01:01/HeV1/bβ2m and Ptal-N*01:01/HeV1/hβ2m were then analyzed. In the Ptal-N*01:01/HeV1/bβ2m structure, residues R3, H31, Q34, D53, W60, Y63 of bβ2m bound to the H chain residues through bottom of the PBG and residues Q8, Y10, R12, N24, Y28, N98, N99 bound to the α3 domain of the H chain. Similar to the Ptal-N*01:01/HeV1/bβ2m complex, in the Ptal-N*01:01/HeV1/hβ2m structure, conserved residues H31, D53, W60, Y63 of hβ2m which correspond to bβ2m, made contact with the bottom of the PBG and conserved residues Q8, Y10, R12, N24 which correspond to bβ2m bound to α3 domain (Figure 2A,2B).
In the overall structures Ptal-N*01:01/HeV1/bβ2m and Ptal-N*01:01/HeV1/hβ2m, the average root-mean-square deviation (RMSD) of residues 1–184 of the H chains (forming the α1α2 PBG) was 0.248 under all Cα atoms superposition (Figure 3A). This finding indicated that there was no difference between these two complexes. The conformations of the similar peptides in the complexes with different β2m were then compared. The structure of the peptide alignment showed that the conformations of HeV1 peptides in these two complexes were quite similar (Figure 3B). In addition, the structures of gp33(KAVYNFATM) presented by H-2Db and complexed with mouse β2m (mβ2m) or hβ2m were aligned. The RMSD of the α1α2 PBG of H-2Db was 0.283 and the overall conformations of peptides in these two structures were also similar (Figure 3C,3D). These data indicate that the β2m substitution between bβ2m and hβ2m, and mβ2m and hβ2m do not affect the conformations of presented peptides.
Sequence alignment showed that the amino acids of β2m from different species are highly conserved (Figure 4). Following analysis of the β2m from different species showed that most of the amino acids of β2m that were forming the hydrogen bonds with the H chain of MHC I were conserved (Figure 4, Table 2). Meanwhile, the diverse residues were also amino acids with similar chemical properties in mammals. However, the key residues involved in β2m binding to the H chain of MHC I showed polymorphisms in chickens, fish and amphibians (Figure 4).
Table 1: The various mammals combine with heterologous β2m. Please click here to download this table.
Table 2: Hydrogen bond interactions between heterologous β2m and heavy chain in MHC I of various species. Please click here to download this table.
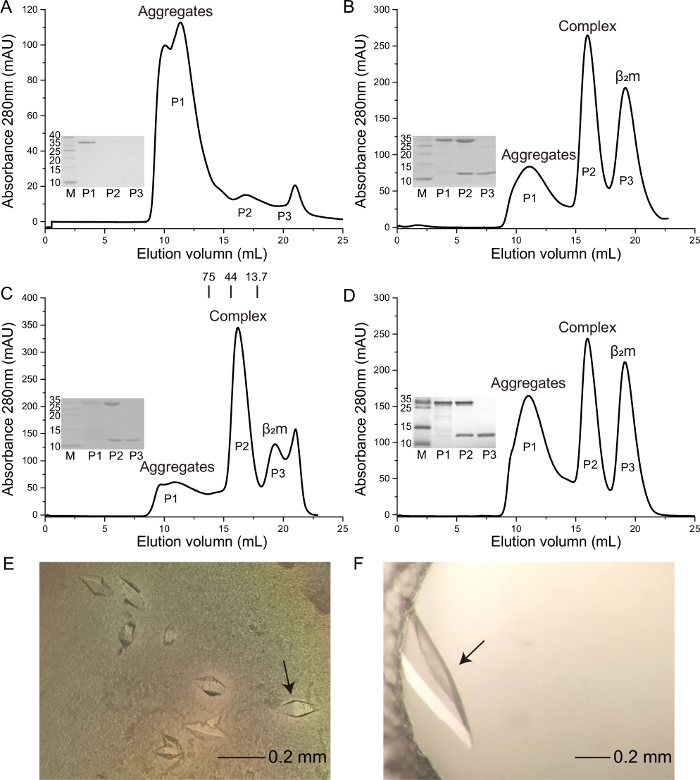
Figure 1: Purification of the soluble and refolded Ptal-N*01:01/HeV1 complex proteins and photographs of the crystal used for diffraction analysis. The M is molecular weight markers in kDa. The P1 is the aggregates. The P2 is the MHC complex. The P3 is the β2m. (A) Ptal-N*01:01/HeV1 complex was not formed without the presence of β2m. (B) H-2Kd complex was formed through renaturation with hβ2m. (C) Ptal-N*01:01/HeV1 complex was formed through renaturation with bβ2m. The profile is marked with the approximate positions of the molecular mass standards of 75.0, 44.0, and 13.7 kDa. (D) Ptal-N*01:01/ HeV1 complex was formed through renaturation with hβ2m. (E) The crystal is formed from Ptal-N*01:01/HeV1 complex, which was formed through renaturation with bβ2m. The black arrow represents the crystal used to collect data during X-ray diffraction. (F) The crystal is formed from Ptal-N*01:01/HeV1 complex which was formed through renaturation with hβ2m. The black arrow represents the crystal used to collect data during X-ray diffraction. Please click here to view a larger version of this figure.
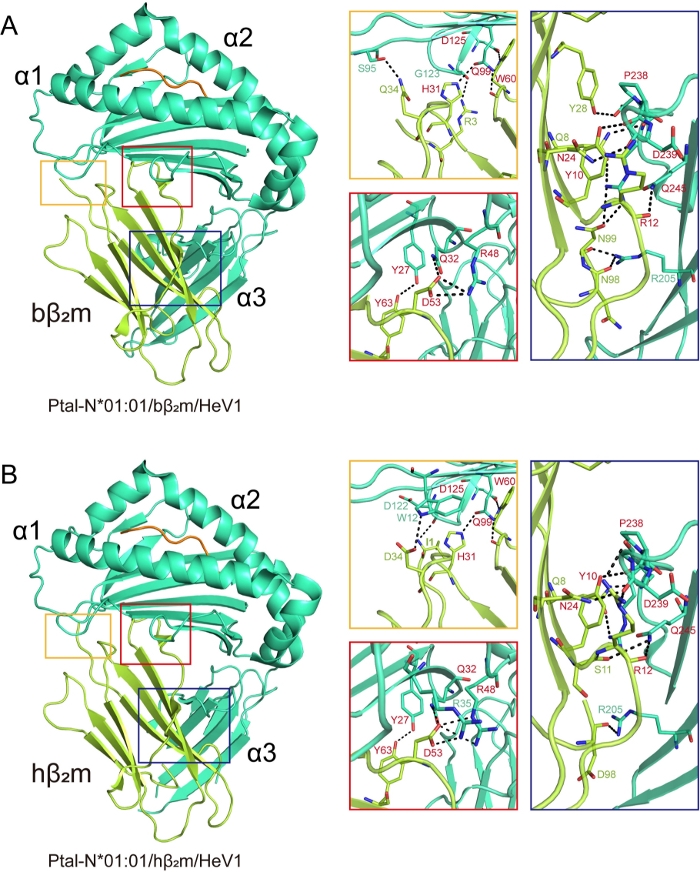
Figure 2: Hydrogen bonding between β2m and heavy chain in hybrid MHC I complexes. Hydrogen bonding between β2m and H chain in (A) Ptal-N*01:01/bβ2m/HeV1 and (B) Ptal-N*01:01/hβ2m/HeV1 MHC complexes. Hydrogen bond interactions are represented by a black dotted line. The square represents the area zoomed in and shown to the right in the corresponding colored boxes. The red represents that the homologous β2m and the heterologous β2m use the same amino acids to form hydrogen bonds with the H chain. Please click here to view a larger version of this figure.
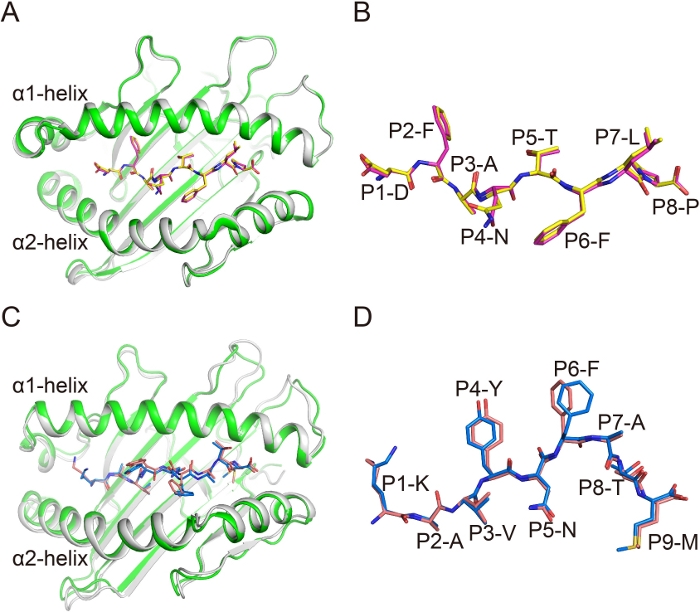
Figure 3: Similar conformation of the MHC complex and the antigenic peptides in hybrid MHC I complexes. (A) The superimposition of α1α2 domains of Ptal-N*01:01/bβ2m (green) and Ptal-N*01:01/hβ2m (gray). (B) The superposition of HeV1 peptide with the superimposition of α1α2 domain of each Ptal-N*01:01 molecule. HeV1 Peptide is represented as pink in Ptal-N*01:01/bβ2m and as yellow in Ptal-N*01:01/hβ2m. (C) The superimposition of α1α2 domains of H-2Db /mouse β2m (mβ2m) (green) and H-2Db /hβ2m (gray). (D) The superposition of gp33 peptide with the superimposition of α1α2 domain of each H-2Db molecule. Peptide gp33 is represented as blue in H-2Db /mβ2m and as pink in H-2Db /hβ2m. Please click here to view a larger version of this figure.
Figure 4: Structure-based sequence alignment of hβ2m with β2m of other species. The black arrows denote β-strands. The residues highlighted in red are completely conserved, and the residues in blue boxes are highly (>80%) conserved. The yellow triangles represent the key amino acids for the interaction between the β2m and H chains. The sequence alignment was generated using Clustal X32 and ESPript33. Please click here to view a larger version of this figure.
