1. Introduction of expression plasmids into competent Pro-auxotrophic E. coli cells
- Mix 1 ng of each sample plasmid, pQE-80L H6-EGFP, pQE-80L H6-NowGFP, and pQE-80L H6-KillerOrange, with 50 µL of chemically competent or electrocompetent cells of the Pro-auxotrophic E. coli K12- derived strain JM83 (Addgene #50348, or ATCC #35607) for transformation by the heat shock method or electroporation according to the protocols available46,47.
NOTE: The expression vector pQE-80L EGFP-H6 encodes a C-terminally 6xHis-tagged enhanced green fluorescent protein (EGFP), the expression vectors pQE-80L H6-NowGFP and pQE-80L H6-KillerOrange encode an N-terminally 6xHis-tagged NowGFP48 or an N-terminally 6xHis-tagged KillerOrange49, respectively, each in a pQE-80L plasmid backbone. All target genes are under the control of a bacterial T5 promoter regulated by the lac operator. Ampicillin resistance was used as a selection marker and colE1 as the origin of replication. See the Table of Materials for further information on the pQE-80L plasmid. Alternative Pro-auxotrophic E. coli originating from strains K-12 and B can be used for expression as well and should yield similar results. The calculated molecular mass of the H6-tagged singly protonated ([M+H]+) wild-type proteins (after chromophore maturation) are 27,745.33 Da (EGFP- H6), 27,931.50 Da (H6-NowGFP), and 27,606.09 Da (H6-KillerOrange). The primary structures of the target proteins are given in Table 1 (His-tag underlined). The glutamine (Q) within the His-tag sequence of NowGFP was identified by DNA sequencing after subcloning of the NowGFP cDNA received from Pletnev et al.51 into the pQE-80L plasmid. It does not impede protein purification or the properties of the fluorescent protein. - Recover the cells in 950 µL of SOC medium at 37 °C for 1 h.
- Spread 50 µL of the recovered cells onto Luria Agar (LA) medium plates (see Supplementary Material) containing glucose (10 g/L) and ampicillin (100 µg/mL).
- Incubate the LA medium plates at 37 °C overnight or 30 °C for 24 h.
2. Production of recombinant wild-type fluorescent proteins (harboring canonical proline) and procedure for selective pressure incorporation (SPI) to produce fluorescent proteins with proline analogs (S-Flp, R-Flp, Dfp, Dhp)
- Overnight culture of Pro-auxotrophic E. coli K12-derived strain JM83 harboring pQE-80L EGFP- H6, pQE-80L H6-NowGFP, and pQE-80L H6-KillerOrange
- Use a sterile pipette tip or inoculation loop to select a single colony from an LA medium plate and resuspend the cells in 5 mL of Lysogeny Broth (LB) medium (see Supplementary Material; containing 10 g/L glucose, 100 µg/mL ampicillin) in a sterile 14 mL polystyrene culture tube.
NOTE: Freshly transformed colonies are recommended for inoculation. Cells on LA medium plates (from step 2.2.) stored at 4 °C should be used within a few days. - Grow the cell culture overnight at 37 °C in an orbital shaker at 200-250 rpm.
- Use a sterile pipette tip or inoculation loop to select a single colony from an LA medium plate and resuspend the cells in 5 mL of Lysogeny Broth (LB) medium (see Supplementary Material; containing 10 g/L glucose, 100 µg/mL ampicillin) in a sterile 14 mL polystyrene culture tube.
- Production of recombinant EGFP- H6, H6-NowGFP and H6-KillerOrange with native proline and the corresponding protein variants with proline analogs
- Inoculate 200 mL of fresh NMM medium (7.5 mM (NH4)2SO4, 50 mM K2HPO4 and 22 mM KH2PO4, 8.5 mM NaCl, 1 mM MgSO4, 20 mM D-glucose, 1 µg/mL FeCl2, 1 µg/mL CaCl2, 10 µg/mL thiamine, 10 µg/mL biotin, 0.01 µg/mL trace elements (CuSO4, ZnCl2, MnCl2, (NH4)2MoO4), pH ~7.2) with all canonical l-amino acids (50 mg/L); see Supplementary Material) supplemented with 100 µg/mL ampicillin with 2 mL of the overnight culture in a 2-L Erlenmeyer flask.
NOTE: Depending on the type of protein, alternative cultivation media like MOPS medium52, glucose-mineral salts medium53, Davis minimal medium54, M9 minimal medium55, or GMML56 can be tested to optimize protein yield. - Incubate the cells at 37 °C in an orbital shaker at 220 rpm for ~3 h 30 min.
- Measure the optical density at 600 nm (OD600) in a spectrophotometer every 30 min until an OD600 value of ~ 0.7 is reached.
NOTE: For OD600 determination in a spectrophotometer, the cuvette should have a path length of 1 cm. The corresponding cultivation medium is used for the reference ("zero") measurement. The incubation time until an OD600 value of ~ 0.7 is reached may depend on culture volume. 3 h 30 min is an approximate value. In a variation of the protocol substeps 2.2.1-2.2.3., the culture from substep 2.1.2. is used to inoculate fresh NMMΔPro medium (see Supplementary Material) supplemented with a limited concentration of proline (e.g., 5 mg/L instead of 50 mg/L), and the cells are grown overnight at 37 °C in an orbital shaker at 220 rpm. The next day, the OD600 determination is performed every 30 min until the differences between the measurements are less than 0.05. The maximum OD600 value should be around 1 (± 0.3). The initial, limited proline concentration can be adjusted depending on the expression strain and cultivation medium (see Discussion). - Spin down the cell suspension for 10 min at 3,000 x g and 4 °C.
- Gently decant the supernatant into waste.
- Washing step: Resuspend the cells in 50 mL of ice-cold NMMΔAA (without any amino acid, see Supplementary Material) or NMMΔPro (without proline, see Supplementary Material) medium by careful pipetting.
NOTE: For this washing step, either of the two stated buffers can be used, since it is only important to get rid of residual proline in the incubation medium. - Separate the cells from the medium by sedimentation at 4 °C in a centrifuge at 3,000 x g for 10 min.
- Gently decant the supernatant into waste.
- Gently pipette up and down to resuspend the cell pellet in 200 mL of NMMΔPro medium supplemented with 100 µg/mL ampicillin into a 2-L Erlenmeyer flask.
NOTE: The resulting cell suspension can be separated into several samples of equal volume to obtain identical start cultures for comparing protein expression in the presence of Pro or proline analogs (e.g., separation into 4 x 50 mL in 100 mL Erlenmeyer flasks). - Incubate for 30 min at 37 °C in an orbital shaker at 220 rpm for the complete depletion of Pro.
NOTE: This step is critical to completely deplete Pro from cells. - Add an appropriate volume of either L-proline or R-Flp, S-Flp, Dfp, and Dhp from 50 mM stock solution to adjust 1 mM final concentration in the cell suspension.
NOTE: As a general rule, fresh 50 mM stock solutions of Pro or proline derivatives should always be prepared prior to use. Only if hydrolysis of the particular canonical or non-canonical amino acid in an aqueous solution is not a concern, frozen stocks may be used as well. - Add 0.5 mM IPTG from a 1 M stock solution to induce target protein expression.
- Express the target protein overnight (12 h) at 37 °C in an orbital shaker at 220 rpm.
- Measure OD600 on the next day.
NOTE: OD600 determination is done to quantify the amount of cells after protein expression. A lower OD600 value compared to the value measured before the washing step 2.2.3 indicates cytotoxicity of the supplied amino acid. In this case, the procedure should be repeated with the concentration of the supplied amino acid minimized (down to 0.1 mM). - Centrifuge and collect the bacterial cells at 5,000 x g and 4 °C for 10 min and decant the supernatant into waste.
- Washing step: Resuspend the cells by careful pipetting in 50 mL of binding buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM DTT) containing 10% glycerol and transfer the cell suspension into a 50 mL conical polystyrene tube.
- Centrifuge and collect the bacterial cells at 5,000 x g and 4 °C for 10 min and decant the supernatant into waste.
- Store the cell pellet in a 50 mL conical polystyrene tube at -20 °C or -80 °C until further use (protein purification, see below).
- Inoculate 200 mL of fresh NMM medium (7.5 mM (NH4)2SO4, 50 mM K2HPO4 and 22 mM KH2PO4, 8.5 mM NaCl, 1 mM MgSO4, 20 mM D-glucose, 1 µg/mL FeCl2, 1 µg/mL CaCl2, 10 µg/mL thiamine, 10 µg/mL biotin, 0.01 µg/mL trace elements (CuSO4, ZnCl2, MnCl2, (NH4)2MoO4), pH ~7.2) with all canonical l-amino acids (50 mg/L); see Supplementary Material) supplemented with 100 µg/mL ampicillin with 2 mL of the overnight culture in a 2-L Erlenmeyer flask.
3. Purification procedure of protein samples by immobilized metal ion affinity chromatography (IMAC)
- Bacterial cell lysis
NOTE: Perform all steps of cell lysis on ice or at 4 °C to prevent degradation of the target protein.- Thaw the bacterial cell pellet on ice or at 4 °C in a 50 mL conical polystyrene tube for 10-20 min.
- Add 10 mL of ice-cold binding buffer (see Supplementary Material) and gently pipette up and down to resuspend the cell pellet.
- Add 100 µL of 50 mg/mL lysozyme, 100 µL of 1 mg/mL DNase I, 100 µL of 1 mg/mL RNase A, 30 µL of 1 M MgCl2. Carefully invert the cell suspension five times, and keep the closed tube on ice or at 4 °C for 60 min.
NOTE: Lysozyme induces chemical cell lysis by disrupting the bacterial cell wall. - Sonicate the sample for cell disruption using an ultrasound homogenizer (e.g., 3 times for 3 min in a 50 mL polystyrene tube on an ice-water mixture, pulse 2 s/pause 4 s, 45% amplitude).
NOTE: Alternatively, other cell disruption methods can be used, e.g., high-pressure homogenization in 20 cycles at 14,000 psi. If necessary, dilute using a binding buffer (see Supplementary Material) to reach the minimal instrument volume. Moreover, protein extraction reagents can be used for cell disruption. See the Table of Materials for examples. - Centrifuge for 60 min at 18,000 x g, 4 °C.
- Note down the liquid volume for substep 3.1.9. and pour the supernatant into a fresh 50 mL polystyrene tube.
- Clear the supernatant using a membrane filter with 0.45 µm pore diameter.
- Take a sample of "lysate" for SDS-PAGE (see section 4. below); this corresponds to "soluble protein fraction", the supernatant from substep 3.1.6.
- Add an equal volume of ddH2O as determined in substep 3.1.6. to resuspend the cell debris in order to maintain the same dilution of the samples for subsequent SDS-PAGE analysis.
- Take a sample of "pellet" for SDS-PAGE (see section 4. below); this corresponds to "insoluble protein fraction", the cell debris suspension from substep 3.1.9.
- Immobilized metal affinity chromatography (IMAC) purification
NOTE: Purification of the target fluorescent protein can be performed at 4 °C or at room temperature (RT). For the latter option, wait for the lysate, column, and all buffers to equilibrate at RT to prevent air bubble formation due to vaporization of air trapped in cold solution upon placement into a warm column.- Purify the sample using a 1 mL prepacked or self-packed IMAC FPLC (fast protein [or performance] liquid chromatography) column according to the manufacturer's instructions; set maximum column pressure to 0.3 MPa, and flow rate to 1 mL/min; use binding buffer (see Supplementary Material) for column equilibration, wash buffer (see Supplementary Material) for the wash step, and elution buffer (see Supplementary Material) for target protein elution.
NOTE: Alternatively, an automated FPLC system can be applied to elute the target protein with elution buffer running a linear imidazole concentration gradient (20-200 mM). - Collect and pool the eluate fractions with fluorescent proteins (choose by visible green or orange color).
- Transfer the pooled fractions into a dialysis membrane (molecular weight cutoff (MWCO) of 5,000-10,000) according to the manufacturer's instructions and dialyze at least three times against dialysis buffer or MS buffer (see Supplementary Material). For instance, perform dialysis of a 1-mL sample three times against 100 mL of buffer for at least 2 h each round. For a detailed protocol, refer to Budisa et al.34.
- Prepare a 1:100-fold dilution of the dialyzed elution fraction in PBS buffer (see Supplementary Material).
- Record the absorbance spectrum of the diluted samples in a UV-Vis spectrophotometer.
- Calculate the protein concentration based on the Lambert-Beer law as follows, using literature values of the molar extinction coefficients ε at specific wavelength (for EGFP at 488 nm ε488 = 55,000 cm-1·M-1, NowGFP at 493 nm ε493 = 53,600 cm-1·M-1, KillerOrange at 514 nm ε514 = 22,600 cm-1·M-1):
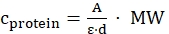 (Lambert-Beer law)
(Lambert-Beer law)
cprotein = protein concentration [mg/mL]
A = absorbance at specific wavelength
ε = molar extinction coefficient at specific wavelength [M-1·cm-1]
d = cuvette path length, here 1 cm
MW = molecular weight of protein [g/mol]
Use dialysis buffer (see Supplementary Material) for the reference ("zero") measurement. - Take a sample of "eluate" for SDS-PAGE (see section 4 below), load 1-10 µg of protein (calculated according to the previous step) per sample well for Coomassie Brilliant Blue-stained gels.
NOTE: Adjust SDS sample amounts depending on the applied staining method and sensitivity of the dye if compounds different from Coomassie Brilliant Blue are used for protein band staining. - Freeze and store the protein sample in dialysis buffer (see Supplementary Material) at -80 °C.
NOTE: Under this storage condition, protein samples should be stable for at least 6 months. Alternative laboratory UV-Vis and fluorescence spectrophotometers can be used for the recording of absorption and fluorescence emission spectra of target proteins. The following excitation wavelengths can be applied for fluorescence emission measurements: 488 nm (EGFP), 493 nm (NowGFP), and 510 nm (KillerOrange).
- Purify the sample using a 1 mL prepacked or self-packed IMAC FPLC (fast protein [or performance] liquid chromatography) column according to the manufacturer's instructions; set maximum column pressure to 0.3 MPa, and flow rate to 1 mL/min; use binding buffer (see Supplementary Material) for column equilibration, wash buffer (see Supplementary Material) for the wash step, and elution buffer (see Supplementary Material) for target protein elution.
4. SDS-PAGE sample preparation
- Determine absorbance A at 280 nm (A280nm) for samples "eluate", "lysate" and "pellet" from section 3. Adjust the probe volume to achieve A280nm = 2 by adding an appropriate amount of elution buffer (final probe volume should be at least 80 µL).
- Mix the sample at a ratio of 4:1 (v/v) with 5x SDS loading dye buffer (see Supplementary Material) by pipetting, e.g., 80 µL of the sample with 20 µL of 5x SDS loading buffer.
- Boil the SDS samples at 95 °C for 5 min in a water bath or heat block to denature the proteins.
- Allow the samples to cool down to RT and spin down the probes at 13,000 x g for 1 min in a microcentrifuge prior to loading onto the gel.
- Use 5-10 µL for Coomassie Brilliant Blue-stained SDS-PAGE. For details of the SDS-PAGE procedure, consult57.
NOTE: Adjust the SDS sample amounts depending on the applied staining method and sensitivity of the dye. The result of the SDS-PAGE should be checked carefully to ensure that the samples contain more than 95% of the total protein in a band corresponding to the expected molecular weight of the desired protein. For this, take a photograph of the Coomassie-stained gel and compare the intensity of the protein band of desired molecular weight with the intensity of all other bands in the lane (if any) by densitometry. For densitometric evaluation of band intensities, the software ImageJ can be used58. To experimentally prove the incorporation of the desired ncAAs into the protein of interest, intact protein mass analysis by high-performance liquid chromatography (HPLC) coupled to electrospray ionization time-of-flight mass spectrometry (LC-ESI-TOF-MS) should be carried out as described59 (see protocol section 5 and Table of Materials therein for exemplary equipment).
5. Fluorescence emission of protein variants
- Prior to the procedure, check the result from the SDS-PAGE experiment to make sure that sample purity is >95% (see the NOTE after step 4.5.)
- Adjust the samples of each purified protein variant to a concentration of 0.3 µM, taking the calculated absorbance value at the appropriate wavelength as a reference (substep 3.2.5.). Ensure that the approximate final sample volume is 200 µL.
- Let the diluted samples equilibrate for 1 h at RT.
- Transfer the samples into a 1-cm quartz cuvette and measure a fluorescence emission spectrum of the samples using a fluorescence spectrometer (see Table of Materials) applying the following excitation wavelengths: 488 nm (for EGFP), 493 nm (for NowGFP), 510 nm (for KillerOrange).
6. Denaturation and refolding of EGFP variants
- Prior to the procedure, check the result from the SDS-PAGE experiment to make sure that sample purity is >95% (see the NOTE after step 4.5.)
- Prepare for each purified protein variant two samples of 2 µL final volume at a concentration of 300 µM (see protein concentration determination in substeps 3.2.4-3.2.6).
- Add 18 µL of 1.11x PBS buffer (see Supplementary Material) containing 8.89 M urea and 5.56 mM DTT to 2 µL of each purified protein variant (to obtain 1x PBS containing 8 M urea and 5 mM DTT) to induce denaturation.
NOTE: For steps 6.4-6.6, process each sample separately. - Incubate the samples for 5 min at 95 °C.
- Dilute the 20 µL samples 100-fold by adding 1980 µL of 1x PBS (see Supplementary Material) containing 5 mM DTT to induce renaturation, yielding 0.3 µM final protein concentration and immediately transfer 200 µL of the samples into a 1-cm quartz cuvette.
NOTE: It is very important to work fast here since renaturation starts immediately. - Insert the quartz cuvette into an appropriate fluorescence spectrometer (see Table of Materials) and monitor protein refolding in the samples by acquiring a fluorescence spectrum every 3 s over 30 min. For each protein variant, use 295 nm fluorescence excitation for the first sample and 488 nm fluorescence excitation for the second one.
- Transfer the refolding samples into 1.5 mL microcentrifuge tubes, close the lid and store the samples at RT in the dark for 24 h to allow complete refolding of EGFP variants.
- Measure fluorescence emission of refolded protein samples according to step 6.6 using the same excitation wavelength as before to capture the temporal endpoint of fluorescence recovery.
At the beginning of the study, we selected three different fluorescent protein variants sharing the parent GFP architecture. The first protein selected was EGFP, which is an engineered variant derived from the original GFP from the jellyfish Aequorea victoria containing Phe64Leu/Ser65Thr mutations. The second selected protein was NowGFP51,60. It is also a variant of A. victoria GFP derived by mutagenesis in several steps via preceding fluorescent proteins. NowGFP contains 18 mutations compared to its immediate predecessor fluorescent protein "Cerulean"61. In turn, the "Cerulean" protein is a derivative of the enhanced cyan fluorescent protein (ECFP)62,63, a protein previously selected by directed laboratory evolution and containing a tryptophan-based chromophore. Both, EGFP and NowGFP are widely used in cell biology and biophysical studies, and they contain ten conserved proline residues in their structures. In addition, NowGFP has an eleventh proline residue at position 230, which appeared due to the extensive mutation history of this protein variant. The third protein selected was the KillerOrange fluorescent protein64,65. It is a derivative of the chromoprotein anm2CP from the hydrozoan genus Anthoathecata. The protein sequence contains 15 proline residues, and the chromophore is based on a tryptophan rather than a tyrosine residue. High-resolution X-ray structures have been reported for all three selected proteins (Figure 2)51,65,66.
In the first step, proline analogs (Figure 1D) were incorporated into all proline positions of three model proteins (EGFP, NowGFP, and KillerOrange) by selective pressure incorporation (SPI, a scheme of the procedure is given in Figure 3). Instrumentally, the proline-auxotrophic E. coli K12 strain JM8367 was used for expression of the proteins in the presence of proline and analogs (Figure 1D), yielding wild-type and modified proteins, respectively. Pellets from cells expressing the native protein and variants bearing S-Flp and Dhp had the typical bright color due to the intact chromophore, whereas variants containing R-Flp and Dfp remained colorless, indicating misfolding and deposition of unfolded protein in inclusion bodies (Figure 4A). SDS-PAGE analysis of the expressed samples verified the presence of insoluble R-Flp-containing proteins (Figure 4B–D), which precluded further investigations. Although this is beyond the scope of the present study, it should be noted that protein solubility and misfolding issues can be alleviated to some extent by in vitro refolding procedures68. In contrast, native proteins as well as S-Flp- and Dhp-bearing variants were found mainly in the soluble fractions (Figure 4B–D). The wild-type, as well as S-Flp- and Dhp-containing variants, could be further isolated and characterized in fluorescence studies. Soluble proteins were purified by immobilized metal ion affinity chromatography (IMAC), yielding 20-30 mg/L of culture volume for EGFP, 60-80 mg for NowGFP and KillerOrange, whose yields for wild-type and modified proteins were very similar. Liquid chromatography-mass spectrometry (LC-MS)-coupled analysis confirmed the expected identity and purity of the isolates obtained in this fashion (Figure 5). In the mass spectra, each proline replacement with S-Flp produced a +18 Da shift per each proline residue in the sequence, while for the proline-to-Dhp replacement, the shift was −2 Da per residue.
In the next step, light absorption and emission spectra were recorded to analyze the potential effects of non-canonical proline analogs incorporation on the spectroscopic properties of the parent fluorescent proteins (Figure 6). UV-Vis absorption spectra showed a typical band around 280 nm characteristic for aromatic residues, tyrosine, and tryptophan, while the chromophore absorbance was found at 488 nm for EGFP, and 493 nm for NowGFP (Figure 6A,B). In KillerOrange, the chromophore absorbance region comprised two bands (Figure 6C), which correspond to two possible configurational and charge states of the complex chromophore. The band around 510 nm is known as the state from which fluorescence occurs with high quantum yield49,65. In the proline replacement variants, the following was observed: Incorporation of Dhp did not change the absorbance spectra of EGFP and NowGFP, while S-Flp produced an enhanced UV absorption. The latter can be explained by induced differences in the tryptophan residue microenvironments, particularly Trp57 sandwiched between three S-Flp in the PVPWP motif (Figure 6A,B)69. A more trivial explanation for a higher UV absorption, however, may stem from an increased fraction of improperly folded protein. Since the concentration of the protein was assessed by quantification of absorbance features, the presence of a protein with an improperly mature chromophore can increase the absorbance, while this fraction is not counted in the overall concentration (Figure 6A,B). Supporting this hypothesis, we observed that the S-Flp-containing EGFP exhibited a markedly reduced ratio of chromophore versus combined tryptophan and tyrosine absorbance (ε(CRO)/ε(Tyr+Trp) = 0.96) as compared to a higher value (1.57) in the parent protein (Table 2)70. The presence of a non-fluorescent fraction in the S-Flp-containing EGFP will be an important contributing factor in further analysis of the protein properties. In the KillerOrange variant containing S-Flp, an enhanced absorbance alongside a red-shift in the chromophore band was observed. This fact indicated that the chromophore formation favored a configuration with a large fluorescence quantum yield (Figure 6C).
Subsequently, we analyzed the fluorescence spectra of the proteins recorded upon excitation at the corresponding maximum absorbance wavelengths. The results show that the spectra remained essentially identical for the examined fluorescent protein variants bearing proline and replacements, S-Flp and Dhp. This outcome implies that the analogs did not alter the chemical environment of the chromophore in any case (Figure 6G–I). Despite this fact, marked differences were seen in the fluorescence spectra of KillerOrange recorded upon excitation at 295 nm, hence upon tryptophan excitation. This experiment tracks fluorescence resonance energy transfer (FRET) or direct excitonic coupling that occurs between the tryptophan side chains and the mature chromophore as both are located at a short distance of not more than 25 Å. For EGFP and NowGFP variants, when the emission spectra were measured using 295 nm excitation, a strong chromophore emission was observed alongside hardly any tryptophan emission (Figure 6D,E). However, the variants containing S-Flp exhibited a slightly larger tryptophan-specific emission. This observation can be linked to an uncounted contribution of the unfolded apoprotein that contains tryptophans but not the mature chromophore. Substantially increased tryptophan-specific emission was seen in KillerOrange, indicating a lack of fluorescence quenching via the expected mechanism of excitation energy transfer or excitonic coupling. The protein variants containing proline and S-Flp exhibited comparable tryptophan emission alongside the favored red-shifted fluorescence feature of a high quantum yield. In contrast, the variant that contained Dhp showed a drastic decrease in chromophore fluorescence intensity, presumably due to minor structural effects (Figure 6F).
Next, we compared the folding properties of the proteins by performing an unfolding/renaturation experiment. Fluorescence emission spectra were recorded in the folded state (protocol section 5), after chemical denaturation and, subsequently, in the process of refolding monitored over a period of 24 h (protocol section 6). The spectra were recorded upon excitation at both relevant wavelengths, 295 nm, and at the maxima of the chromophores' absorbance spectra, while the resulting fluorescence is presented as normalized to the maximum value for each protein (Figure 7). At the end of the protocol, we observed that EGFP variants could refold, while the NowGFP and KillerOrange variants – once denatured – remained unfolded (data not shown). Thus, refolding capacities of the original fluorescence proteins varied substantially. Of note, KillerOrange has been developed as a photosensitizer starting from the hydrozoan chromoprotein variant KillerRed65,71, and its refolding typically lags behind in spite of the robust β-barrel structure. In our experiments, we found that the wild-type EGFP chromophore fluorescence recovered only partially, although the tryptophan-specific fluorescence was larger after renaturation (Figure 7A,D). Essentially similar behavior was observed in the variant containing Dhp (Figure 7C,F). In S-Flp-containing EGFP, a similar result was observed when the excitation was performed at the tryptophan-specific wavelength of 295 nm (Figure 7B). Strikingly, the fluorescence recovered to a much higher extend when the chromophore was excited at 488 nm (Figure 7E). It seems that S-Flp induces a much better yield of refolding compared to the other two variants. However, this beneficial effect was not seen when using 295 nm excitation due to unknown molecular interactions.
Subsequently, refolding velocity was monitored by recording fluorescence of both tryptophan, and the chromophore, separately, while the endpoint of the process was determined at 24 h after the start of renaturation. Only EGFP variants showed a relatively fast refolding kinetics that could be evaluated reliably, while none of the denatured NowGFP and KillerOrange variants could recover to a value that enabled further quantitative measurements. In EGFP, tryptophan emission recovery was twice as fast (completed in 750 s) compared to the recovery of chromophore emission (completed in 1,500 s), indicating the complexity of the underlying processes (Figure 8). At both excitation wavelengths, the refolding rate was elevated by the presence of S-Flp, in agreement with literature data25. At the same time, the Dhp-containing variant showed a refolding profile similar to wild-type.
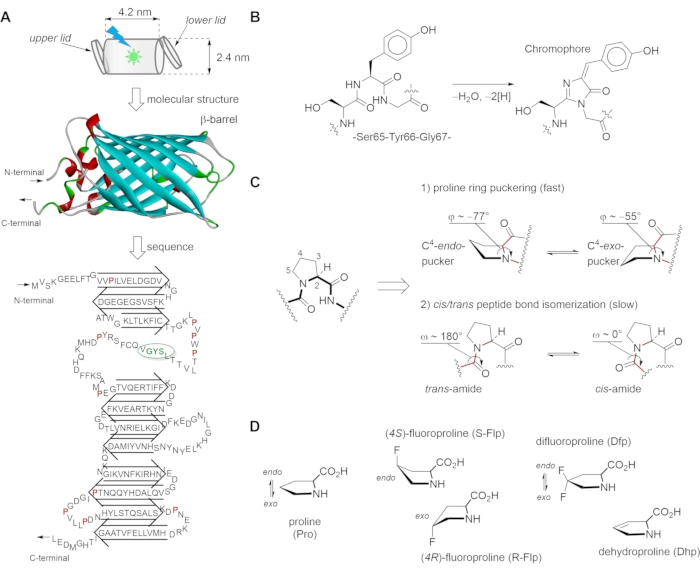
Figure 1: Green fluorescent protein (GFP) structural scaffold, chromophore building, proline conformational transitions and synthetic analogs used in this study. (A) The structure of GFP consists of the β-strands forming a nearly perfect barrel (i.e., a "can" with dimensions 4.2 nm x 2.4 nm) that is capped at both ends by α-helical lids. The 27 kDa GFP protein shows a tertiary structure consisting of eleven β-strands, two short α-helices, and the chromophore in the middle. The conformational states of adjacent prolines are linked to chromophore formation. (B) Autocatalytic maturation (condensation) of the chromophore occurs at residues Ser65, Tyr66, and Gly67, and proceeds in several steps: First, torsional adjustments in the polypeptide backbone to bring the carboxyl carbon of Thr65 into proximity to the amide nitrogen of Gly67. Then, the formation of a heterocyclic imidazoline-5-one ring system occurs upon nucleophilic attack on this carbon atom by the amide nitrogen of glycine and subsequent dehydration. Finally, the system gains visible fluorescence when oxidation of the tyrosine alpha-beta carbon bond by molecular oxygen leads to the extension of the conjugated system of the imidazoline ring system, at the end including the tyrosine phenyl ring and its para-oxygen substituent. The resulting para-hydroxybenzylidene imidazolinone chromophore in the center of the β-barrel is completely separated from the bulk solvent. (C) The skeletal structure formulas and geometries of 1) the proline ring (puckers) and 2) the preceding amide bond represents the main conformational transitions of the proline residue. (D) The proline analogs used in this work with the designated proline ring puckers. The figure was generated using ChemDraw and Discovery Studio Visualizer. The GFP structure is from PDB structure entry 2Q6P. Please click here to view a larger version of this figure.
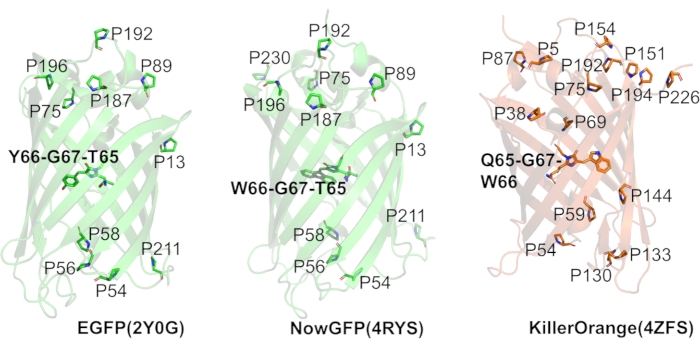
Figure 2: Fluorescent proteins used in this study. The panels show the ribbon representation of the typical β-barrel structures of three different variants of fluorescent proteins: EGFP, NowGFP, and KillerOrange, with ribbon color representing the color of fluorescence emission of each variant. Proline residues (one-letter code) are highlighted as sticks, and the appropriate positions are annotated. Chromophores are shown with initial amino acid composition in bold. All structure representations were produced with PyMol based on the following PDB structure entries: 2Q6P for EGFP, 4RYS for NowGFP, 4ZFS for KillerOrange. Please click here to view a larger version of this figure.
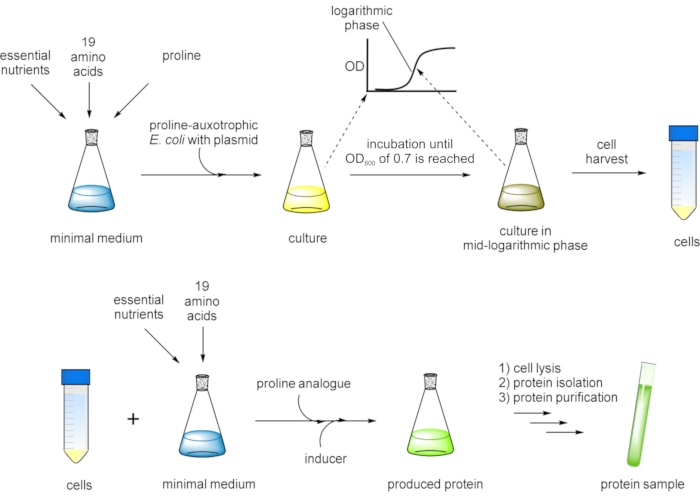
Figure 3: Flow chart presentation of the SPI method for residue-specific incorporation of non-canonical proline analogs. A proline-auxotrophic Escherichia coli (E. coli) host strain carrying the gene of interest on an expression plasmid is grown in a defined minimal medium with all 20 canonical amino acids until an OD600 of ~0.7 is reached at which the cell culture is in the mid-logarithmic growth phase. Cells are harvested and transferred into fresh minimal medium containing 19 canonical amino acids and a proline analog. After the addition of an inducer, protein expression is performed overnight. Finally, the target protein is isolated by cell lysis and purified prior to further analysis. In a variation of the protocol, the cells are grown in a defined minimal medium with 19 canonical amino acids, and proline is added in a limited amount (e.g., one-fifth of the concentration of the other amino acids). By this measure, the cells exhaust proline in the medium before they can exit the logarithmic growth phase, and then, subsequently, the analog is added, and the protein of interest production is induced. Please click here to view a larger version of this figure.
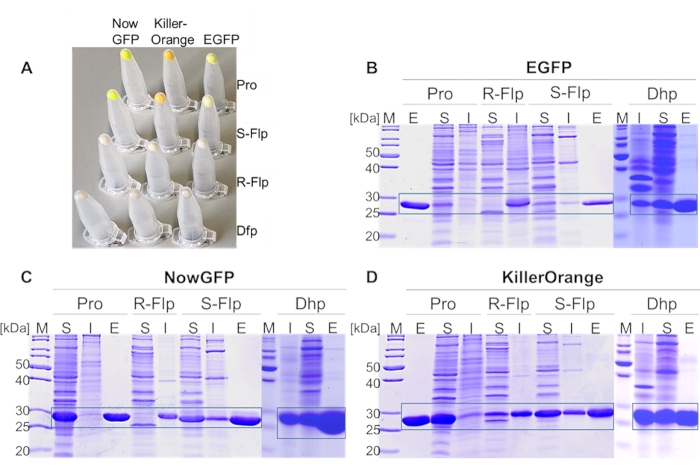
Figure 4: Expression analysis of EGFP, NowGFP, and KillerOrange variants. (A) Cell pellets from 1 mL of expression culture, normalized to OD600 = 2. SDS-PAGE analysis of (B) EGFP, (C) NowGFP, and (D) KillerOrange variants. Soluble (S) and insoluble fractions (I) of each fluorescent protein derivates were loaded on 15% acrylamide gel, as well as eluted fractions (E) from IMAC of soluble proteins. PageRuler Unstained Protein Ladder was used as a marker (M) in the lanes denoted by (M). The expected regions of the particular protein are framed. Incorporated amino acids at proline positions are Pro, R-Flp, S-Flp, and Dhp (in (A) cell pellets from fluorescent protein variants incorporating Dfp instead of Dhp are shown). Gels were stained by 1% (w/v) Coomassie Brillant Blue. Please click here to view a larger version of this figure.
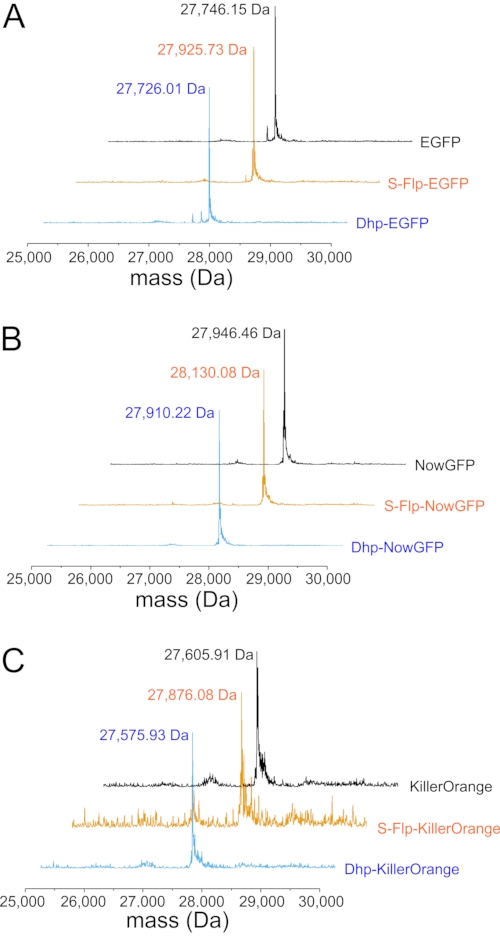
Figure 5: Mass spectrometric analysis of fluorescent protein variants. (A) Representative deconvoluted ESI-MS spectra of H6-tagged EGFP (black), S-Flp-EGFP (orange), and Dhp-EGFP (cyan) with the location of the main mass peaks provided as numbers (in Da). The calculated molecular masses [M+H]+ of the H6-tagged proteins are: For EGFP 27,745.33 Da (observed 27,746,15 Da); for S-Flp-EGFP 27,925.33 Da (observed 27,925.73 Da); for Dhp-EGFP 27,725.33 Da (observed 27,726.01 Da). (B) Representative deconvoluted ESI-MS spectra of H6-tagged NowGFP (black), S-Flp-NowGFP (orange), and Dhp-NowGFP (cyan) with the location of the main mass peaks provided as numbers (in Da). The calculated masses of the H6-tagged proteins are: For NowGFP 27,931.50 Da (observed 27,946.46 Da; the difference of ~16 Da is probably due to oxidation of a methionine in the protein); for S-Flp-NowGFP 28,129.50 Da (observed 28,130.08 Da); for Dhp-NowGFP 27,909.50 Da (observed 27,910.22 Da). (C) Representative deconvoluted ESI-MS spectra of H6-tagged KillerOrange (black), S-Flp-KillerOrange (orange), and Dhp-KillerOrange (cyan) with the location of the main mass peaks provided as numbers (in Da). The calculated masses of the H6-tagged proteins are: For KillerOrange 27,606.09 Da (observed 27,605.91 Da); for S-Flp-KillerOrange 27,876.09 Da (observed 27,876.08 Da); for Dhp-KillerOrange 27,576.09 Da (observed 27,575.93 Da). Deviations between the observed and calculated molecular masses of about 1 Da are within the error range of the ESI-MS equipment. Please click here to view a larger version of this figure.
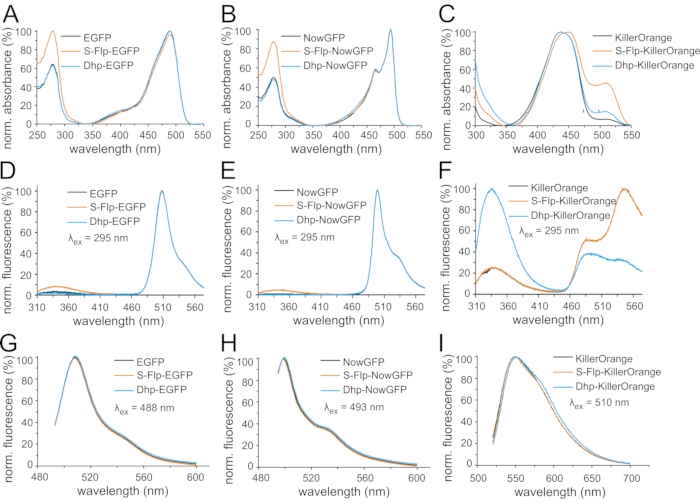
Figure 6: Light absorption and fluorescence emission spectra of fluorescent protein variants. Normalized UV-Vis absorption spectra are shown for the variants (A) of EGFP, (B) of NowGFP, and (C) of KillerOrange. Spectra were normalized to the maximum of chromophore absorbance (around 500 nm). Normalized fluorescence emission spectra are shown of the variants (D,G) of EGFP, (E,H) of NowGFP, and (F,I) of KillerOrange. Spectra in (D,E,F) were measured upon excitation with ultraviolet light (295 nm), for the spectra in (G,H,I) 488 nm, 493 nm, and 510 nm light were used for excitation, respectively, and the spectra were normalized to the respective maxima of chromophore emission (around 500 nm). In each panel, black curves correspond to the spectra of the fluorescent protein variant with native proline, orange curves indicate the spectra of S-Flp-substituted proteins, and blue curves correspond to Dhp-substituted proteins. Please click here to view a larger version of this figure.
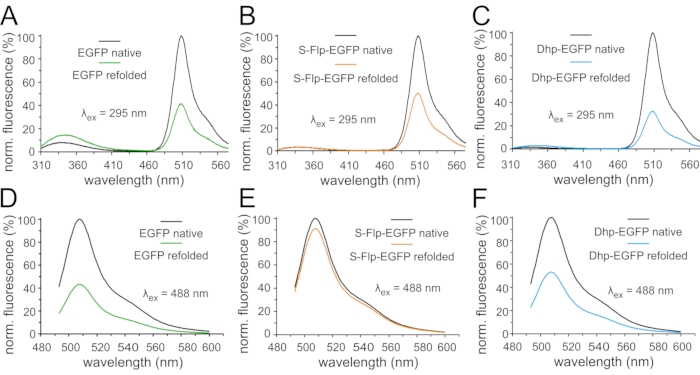
Figure 7: Fluorescence emission spectra of EGFP variants in refolding experiments. Normalized fluorescence emission spectra of 0.3 µM solutions of fluorescent protein variants in the native state and after denaturation and refolding: Spectra in (A,B,C) were measured upon excitation with ultraviolet light (295 nm) (A) for EGFP, (B) for S-Flp-EGFP, and (C) for Dhp-EGFP. Spectra in (D,E,F) were measured upon excitation with green light (488 nm) (D) for EFGP, (E) for S-Flp-EGFP, and (F) for Dhp-EGFP. The emission spectra of the native (black curves) and refolded samples (green corresponds to EGFP, orange to S-Flp-EGFP and blue to Dhp-EGFP, respectively) of each protein variant are normalized to the maximum fluorescence of the appropriate native state. Please click here to view a larger version of this figure.

Figure 8: Monitoring protein folding and chromophore maturation of EGFP variants with fluorescence. (A) Fluorescence emission in the region of Trp fluorescence (emission was set to 330 nm) recorded upon excitation with ultraviolet light (295 nm). (B) Development of the fluorescence amplitude in the region of chromophore emission upon excitation with green light (488 nm). The time-dependent fluorescence traces were normalized to unity (100%) according to the fluorescence amplitude reached at the end of the monitoring interval. In each panel, black curves correspond to the spectra of the fluorescent protein variant with native proline, orange curves indicate the spectra of S-Flp-substituted proteins and blue curves correspond to Dhp-substituted proteins. Please click here to view a larger version of this figure.
| Construct | Amino acid sequences (6xHis tag underlined): | |||
| EGFP-H6 | MVSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVP WPTLVTTLTYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTR AEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVN FKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDH MVLLEFVTAAGITLGMDELYKHHHHHH |
|||
| H6-NowGFP | MRGSHHQHHHGSVSKGEKLFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGK MSLKFICTTGKLPVPWPTLKTTLTWGMQCFARYPDHMKQHDFFKSAMPEGY VQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGVDFKEDGNILGHKLEYN AISGNANITADKQKNGIKAYFTIRHDVEDGSVLLADHYQQNTPIGDGPVLLPD NHYLSTQSKQSKDPNEKRDHMVLLEFVTAAGIPLGADELYK |
|||
| H6-KillerOrange | MRGSHHHHHHGSECGPALFQSDMTFKIFIDGEVNGQKFTIVADGSSKFPH GDFNVHAVCETGKLPMSWKPICHLIQWGEPFFARYPDGISHFAQECFPEG LSIDRTVRFENDGTMTSHHTYELSDTCVVSRITVNCDGFQPDGPIMRDQ LVDILPSETHMFPHGPNAVRQLAFIGFTTADGGLMMGHLDSKMTFNGSR AIEIPGPHFVTIITKQMRDTSDKRDHVCQREVAHAHSVPRITSAIGSDQD |
|||
Table 1: Primary structures of the target proteins. His-tags are underlined in each sequence.
| λ [nm] | ε [M-1·cm-1] (EGFP) | ε [M-1·cm-1] (S-Flp-EGFP) | ε [M-1·cm-1] (Dhp-EGFP) |
| 488 (≡ CRO) | 31,657 (± 1,341) | 22,950 (± 290) | 27,800 (± 542) |
| 280 (≡ Tyr+Trp) | 20,116 (± 172) | 23,800 (± 715) | 17,300 (± 554) |
| Values for extinction coefficient ε (in M-1·cm-1) are calculated from recorded UV-Vis absorption spectra of appropriate EGFP variants using known protein concentrations. Selected wavelength at 280 nm corresponds to the maximum absorbance of aromatic residues, tyrosine and tryptophan, and 488 nm represents the chromophore absorbance wavelength. | |||
Table 2: Extinction coefficients (ε) of EGFP variants at selected wavelengths. Values for the extinction coefficient ε (in M-1·cm-1) are calculated from recorded UV-Vis absorption spectra of appropriate EGFP variants using known protein concentrations. The selected wavelength of 280 nm corresponds to the maximum absorbance of aromatic residues, tyrosine, and tryptophan, whereas 488 nm represents the maximum chromophore absorbance wavelength.
Supplementary Material: Preparation of stock solutions and buffers Please click here to download this File.
