This protocol was successfully used to generate a targeted binary expression reporter system specific for bnl expressing cells5. The cis-regulatory elements (CREs) that control complex spatiotemporal bnl expression are not characterized. Therefore, to achieve spatiotemporal expression under the control of the endogenous bnl regulatory sequence, only the first coding exon of bnl was designed to be replaced with the sequence of the bacterial LexA transactivator. An improved nls-LexA::p65 cassette known to provide optimal expression in Drosophila was selected.
The replacement strategy aimed to generate a chimeric nls-LexA:p65-bnl mRNA under endogenous transcriptional and post-transcriptional control. This chimeric mRNA contained an intact bnl 5' UTR, part of the 5' end and the 3' end of the edited bnl coding exon (first exon), and the complete downstream bnl introns and exons. To preserve the bnl RNA-specific splicing of the replaced exon, small 5' and 3' ends of the replaced coding exon were retained. However, to prevent synthesis of a chimeric Bnl protein fused to nls-LexA:p65, a T2A self-cleaving peptide sequence was incorporated between the residual 5' bnl coding region and the ATG of nls-LexA:p65. Also, a translation stop codon was added after the nls-LexA:p65 sequence to avoid co-translation of a truncated Bnl protein5 (Figure 4 and Figure 5A).
A replacement donor containing all these features was used, which had the T2A–nls-LexA:p65 sequence and 2 kb 5'- and 1.8 kb 3'- homology arms. Long homology arms were used for efficient HDR and insertion of a large DNA fragment at the site of repair (T2A-nls-LexA:p65 is 1.8 kb) (Figure 5A).
Two gRNAs were used to create DSBs at the defined positions to delete most of the first coding exon of bnl. Two gRNAs that matched all criteria described in section 1.3 (PAM sequence underlined) were:
gRNA1: TGTATCTGCGATGCCCCTCATGG
gRNA2: ATCCTTCAGATATTGCGGGATGG
Both gRNA recognition sites were disrupted in the replacement donor by the exogenous T2A–nls-LexA:p65 sequence, which is the easiest way to avoid retargeting of the gRNAs to the engineered locus. The disrupted gRNA recognition sequences in the replacement donor (the exogenous sequences that disrupted gRNA recognition sites in italics) were:
gRNA1: TGTATCTGCG-GGCTCCGGCGAAGGAC…
gRNA2: …AAAAACTCGTTTAGA-CGGGATGG
The pCFD4 gRNA expression vector containing these gRNAs, along with the replacement donor, was co-injected into the germline of nos-Cas9 embryos. Subsequently, the protocol described here was followed to screen for the intended replacement (Figure 5B, C). About 67% of injected G0 animals were fertile. And 56% of the fertile G0s were the founders, which gave rise to HDR-positive progeny. Among the F1 progeny checked, about 23% were positive for successful HDR, and 73% of the positive HDR were "ends-out" (Table 2). Since the first coding exon of bnl was "knocked out", all the HDR lines were found to be homozygous lethal, and the stocks were maintained over balancers.
The generation of a chimeric LexA-bnl mRNA in the cells was validated by RT-PCR analyses (Figure 5D). To verify if the obtained bnl–LexA lines were expressed in the endogenous bnl expression pattern, each "ends-out" HDR line was crossed to LexO-mCherryCAAX transgenic flies5, and the reporter expression pattern was examined in the progeny embryos and larvae. 42 out of 46 lines showed accurate spatiotemporal expression consistent with the previously reported bnl patterns5 (Figure 6). Four lines showed either weak or non-specific expression patterns. We predict that these lines might have accumulated mutations, which we need to verify with thorough sequencing analyses. Together these results confirmed that the HDR-mediated exon replacement strategy could successfully be employed to generate a targeted binary expression system for a gene. The tool can successfully be used to express reporter or ectopic genes in the spatiotemporal patterns of the bnl gene.
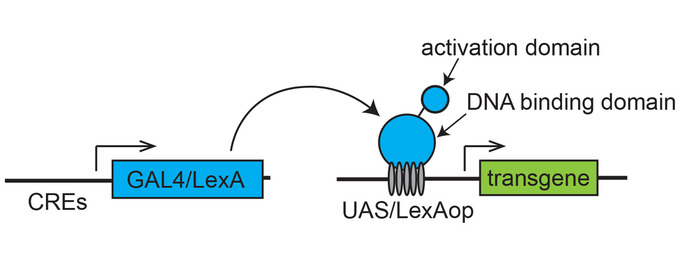
Figure 1: Scheme of a binary transcription system for targeted gene expression. A transactivator (GAL4 or LexA), expressed under the control of cis-regulatory elements (CREs) of a gene, drives an effector transgene placed downstream of the specific transcription binding sites (UAS for Gal4 or LexAop for LexA). Please click here to view a larger version of this figure.
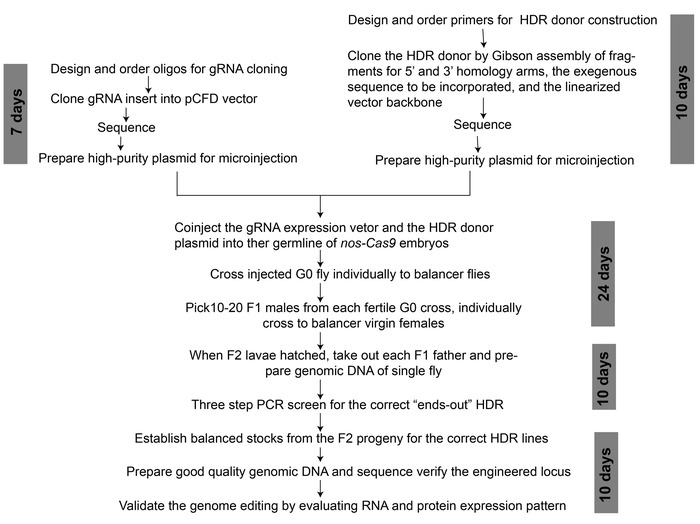
Figure 2: An overview of workflow for CRISPR/Cas9-mediated genome editing to generate a binary transcription system. The approximate time duration required for each step is indicated. Please click here to view a larger version of this figure.
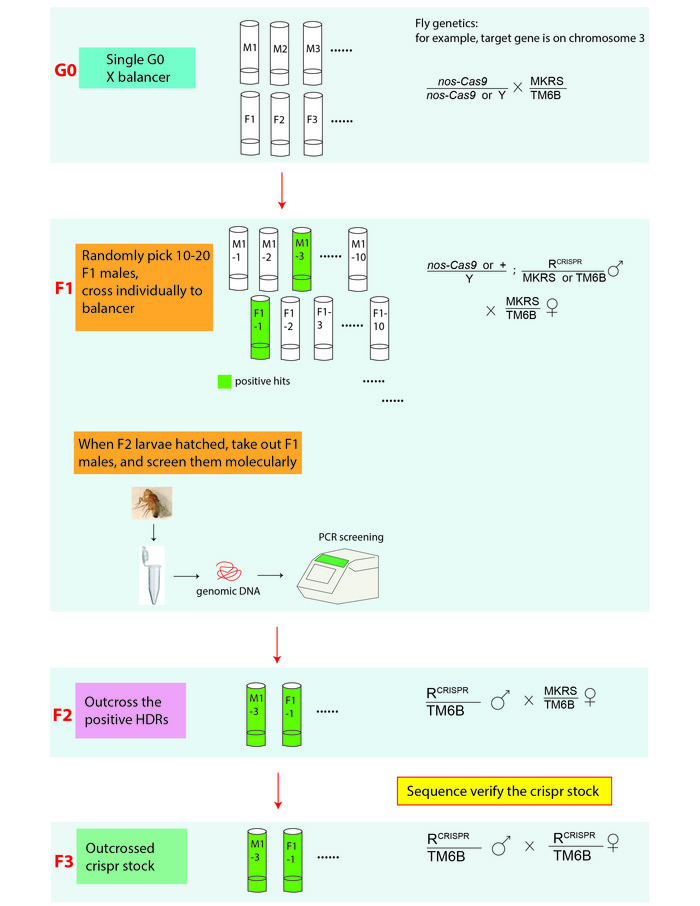
Figure 3: An illustration of the CRISPR screening and genetic cross scheme for establishing genome-edited fly lines. In genetic crosses, R stands for the edited allele that is on the 3rd chromosome. MKRS (Tp(3;3)MRS, M(3)76A[1] kar[1] ry[2] Sb[1]), is a 3rd chromosome marker; TM6B (In(3LR)TM6B, Antp[Hu] e[1] Tb[1]) is a 3rd chromosome balancer. Genome edited fly stocks are verified by PCR amplifying the target regions of interest from the genomic DNA extracted from either the F2 or F3 generation flies and sequence determining the PCR products. Please click here to view a larger version of this figure.
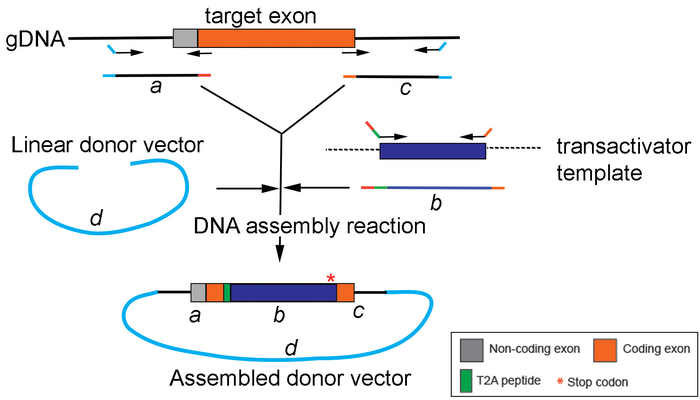
Figure 4: Generation of the replacement cassette by DNA assembly. A schematic drawing depicting PCR amplification and assembly of different PCR products (a, b, and c) into the donor vector (d) using a DNA assembly method. Please click here to view a larger version of this figure.
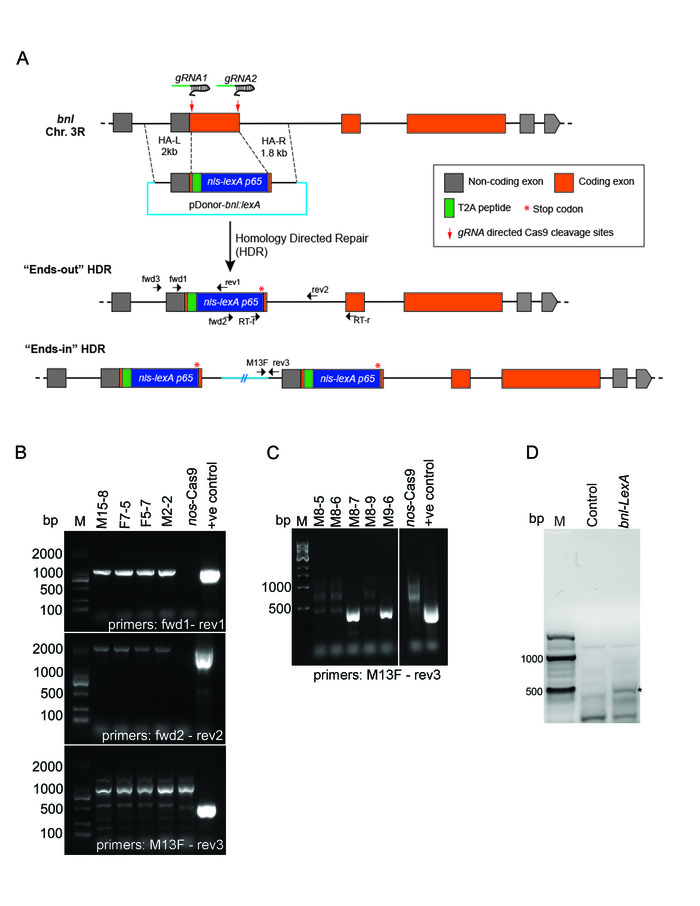
Figure 5: Generation of bnl-LexA by CRISPR/Cas9-mediated exon replacement. (A) Schematic drawing depicting the strategy of the CRISPR/Cas9 mediated HDR for exon replacement in the bnl locus. Box – exon; line- intron; replacement donor (pDonor-bnl:LexA) and two possible outcomes of the HDR were shown. The pDonor-bnl:LexA had the following features: (1)T2A-nls-LexA:p65 (~1.8kb) sequence flanked by 2 kb and 1.8 kb long homology arms (dashed lines), (2) a T2A self-cleaving peptide between the residual N terminal bnl exon and the nls-LexA:p65, and (3) a translation stop codon (red *) after the nls-LexA:p65 sequence. The HDR product retained all the transcriptional and post-transcriptional control of bnl,and the LexA:p65 protein is expected to be produced in the same pattern as endogenous Bnl. Small black arrows show the relative binding sites (not in scale) of the PCR primers (Table 1) used for 3-step screening or RT-PCR validation. (B) Agarose gel pictures showing results of the 3-step PCR screening. PCR products amplified from the genomic DNA of four successful ends-out HDR lines are shown; negative control, the genomic DNA of nos-Cas9 parental line; positive control, pDonor-bnl:LexA plasmid; M, Marker (SL2K DNA ladder). (C) An example of the screening gel showing the expected PCR product using primers M13F and rev3 for ends-in lines; M8-7 and M9-6 are two ends-in lines; negative and positive controls, the same as in B; M, Marker (NEB 1 kb DNA ladder). (D) RT-PCR analysis on total RNA from bnl-LexA and the nos-Cas9 control flies. Forward primer binds to a LexA specific region, reverse primer binds to a downstream bnl exon region; ~440 bp (base-pair) amplification band (*) was detected from RT-PCR on bnl-LexA mRNA, but not from the control RNA. M, 100 bp Marker (NEB). Adapted from Figures 2 and S1 in Du et al. 20175. Please click here to view a larger version of this figure.

Figure 6: Validation of tissue-specific and conditional bnl-LexA expression in different tissues. (A-D) bnl expression (red) in different larval tissues, with the btl expressing cells shown in green. (A,A') Wing disc bnl source ahead of the growing air sac primordium (ASP). (B, C) bnl expression in TR5 transverse connective (TC) (B) and TR2 dorsal branch (DB) (C). (D) bnl expression in genital discs. (E-J) Dynamic bnl expression (red) during embryonic tracheal branch (green) patterning; Small arrows, five bnl sources surrounding the tracheal placode at stage 10. Genotypes: A-J; btl-Gal4, UAS-CD8GFP/+; bnl-LexA,LexO-CherryCAAX/+. (K-L") Hypoxia–induced bnl-LexA expression profile in wing discs and associated TR2 tracheal metamere (TC, DB, dorsal trunk-DT). Genotype: bnl-LexA,LexO-CherryCAAX/+. (K) control discs from ex vivo cultured organs without CoCl2. (L-L") wing discs (L, L') and trachea (DT, (L'')) from cultured ex vivo organs with CoCl2 induced hypoxia. Star, ectopic expression induced by hypoxia. Scale bars = 30 µm; 50 µm (K-L"). Adapted from Figure 3 in Du et al. 20175. Please click here to view a larger version of this figure.
| A. gRNA cloning and sequencing | |
| bnl-lexA gRNA fwd | TATATAGGAAAGATATCCGGGTGAACTTCgTGTATCTGCGAT GCCCCTCAGTTTTAGAGCTAGAAATAGCAAG |
| bnl-lexA gRNA rev | ATTTTAACTTGCTATTTCTAGCTCTAAAACTCCCGCAATATCTGAAGG ATcGACGTTAAATTGAAAATAGGTC |
| T3 primer used for sequencing | CAATTA ACCCTCACTAAAGG-3' |
| Note: nucleotides underlined anneal to U6 promoter or gRNA core on pCFD4 vector, the lowercase g/c was added to aid U6 promoter-dependent transcription | |
| B. HDR donor construction | |
| bnl N-F_pUC19 | AATTCGAGCTCGGTACtgtggtctttgaggctggaac |
| bnl-lexA-N-R | tCCGcaagtCagtAGgctgccgcgtccttcgccggaGCCCGCAGATACAAGGCCC C |
| lexA-F | CTactGacttgCGGaGAtGTcGAaGAGAACCCtGGCCCtATGCCACCCAA GAAGAAGC |
| lexA-R | CTAAACGAGTTTTTAAGCAAACTCACTC |
| bnl lexA-C Fwd | TAAAAACTCGTTTAGACGGGATGGCGTTGTCAAC |
| bnl C-R_pUC19 | GCCAAGCTTGCATGCCtcgcataattgccgcctgg |
| Note: nucleotides in capital overlap with pUC19 vector for Gibson Assembly, nucleotides underlined were sequence overhang for T2A peptide addition. | |
| C. HDR screening and sequencing | |
| bnl-lexA scr fwd1 | GTGGCGCACGCCCAATAAAC |
| bnl-lexA scr rev1 | GATCCCAGCCAATCTCCGTTG |
| bnl-lexA scr fwd2 | CAACGGAGATTGGCTGGGATC |
| bnl-lexA scr rev2 | CTGGCCAACTGTAGGGAAGTC |
| ends-in check rev3 | GCAATGTTATGCAATGCGTTGAC |
| bnl-lexA seq fwd3 | CACTTGTCGCCCATATTGATACAATTG |
| NOTE: These primers were used for PCR screening and sequencing, the approximate locations of primer binding sites are shown in Figure 5A as fwd1-2 and rev1-3. | |
| D. RT-PCR analysis | |
| RT-f | GATATGGATTTCTCCGCTTTGCTG |
| RT-r | CCATGCAGAGATACAGGCAAGTG |
Table 1: Primers used in this study. (A) Primers for cloning gRNA expression vector and sequencing. (B) Primers for cloning HDR donor template. (C) Primers for screening and sequencing of the HDR products. (D) Primers used for RT-PCR verification of the chimeric LexA-bnl mRNA product.
| genotype | HDR transmission rate % (# HDR-yielding G0/# fertile G0) | # HDR-positive F1/# total F1 checked (%) | # "ends-out" HDR/# total HDR (%) |
| bnl-LexA | 56 (15/27) | 23 (60/259) | 73 (46/60) |
Table 2: The efficiency of CRISPR/Cas9-mediated HDR. The HDR transmission rate is calculated as the # of HDR-yielding G0/# of total fertile G0. Successful HDR% is calculated as the # of HDR-positive F1/# of total F1 screened. "Ends-out"% is calculated as the # of "ends-out" HDR/# of total positive HDR.
