A significant challenge in characterizing the effect of molecular perturbations on C. elegans embryonic development is that it takes about 10 h for embryos to progress from first cleavage to the end of elongation at 20°16. A semi-high-throughput method in which large cohorts of embryos can be simultaneously imaged is useful for events on this time-scale because it permits imaging of multiple conditions in parallel with a sufficient ensemble size for each condition to enable quantitative analysis (Figure 1A).
Semi-high throughput imaging method and multi-marker strains
The semi-high-throughput imaging method described here (outlined in Figure 1B), employs a 384-well glass bottom plate; the multi-well format allows for up to 14 conditions to be arrayed in parallel and the small size of the wells constrains the search area, simplifying the identification of fields containing embryos for high resolution imaging. Here, a confocal scanner box was used, equipped with a microlens-enhanced dual Nipkow spinning disk, a 512 x 512 EM-CCD camera, a high-precision auto-XY-Stage (designated resolution 0.1 µm) and motorized z-axis control (designated resolution 0.1 µm). However, the protocol can be adapted for any confocal microscope with precision point-visiting capabilities and a stage adaptor that can accommodate a multi-well plate. A significant challenge in developing this method was devising a means to generate a plate of embryos at the same developmental stage so that the entirety of development can be captured for all of the embryos in each field. This is a challenge because the embryos arrayed onto the imaging plate first will continue to develop while later samples are dissected, resulting in a gradient of staging across the wells. To circumvent this issue, early stage embryos (2-8 cell stage) are selected and keep the dissection media and imaging plate on ice; this stalls embryogenesis for the dissected embryos until all conditions have been arrayed without significantly impacting embryonic viability10. To limit the time that the embryos sit on ice to 1 hour, two researchers perform the dissections simultaneously, which allows for the set-up of 14 different conditions in about 50 min. After dissection, the plate is spun at 600 x g for 1 min to sediment the embryos and wells are pre-scanned at low magnification to locate fields with groups of suitable embryos for high-magnification imaging; point visit locations are marked. Embryos are filmed in a temperature-controlled room overnight using a 60x oil immersion 1.35 NA lens. The samples are configured in a 4 well by 4 well region so that the surface area of the plate that is used in an experiment is comparable to a 22 x 22 mm2 standard coverslip, which allows for the use of a 60x oil objective. Uniform spreading of oil across this region prior to the start of imaging is critical for successful long-term image acquisition. The developing embryos are imaged in a solution containing anesthetic; this ensures that when the older embryos within the well hatch, their movement does not disrupt the position of adjacent younger embryos that are being imaged. Due to the impenetrable nature of the eggshell, the anesthetic does not affect movement prior to hatching. Under these conditions, 3D time-lapse data is collected in 3-channels for a total of 80-100 embryos in a single overnight experiment (discussed more below).
C. elegans embryonic development occurs in two phases. First, 10 rounds of cell division coupled to cell fate specification generate the three germ layers (ectoderm, mesoderm, and endoderm) over a 6 hour period17. In the following 7 h, morphogenetic events drive the formation of differentiated tissues8,16. To capture these two aspects of development, we built a pair of custom strains, the Germ Layer and Morphogenesis strains10 (Figure 2A), and used the semi-high-throughput protocol to image embryos from both strains. The Germ Layer strain expresses transgene-encoded fluorescent proteins that mark nuclei in the ectoderm, mesoderm and endoderm in red, yellow, and green. This strain contains three reporter transgenes: (1) a transgene expressing PHA-4::GFP that marks nuclei in the intestine and pharynx (green endoderm)10,18,19, (2) a transgene expressing an mCherry-histone fusion in the epidermis and in ~1/3 of neurons (red ectoderm; Figure 2A), and (3) a transgene that simultaneously expresses mCherry and GFP-tagged histones in body wall muscle (yellow mesoderm). The Morphogenesis strain has two transgenes that express: (1) a green epithelial junction marker (DLG-1::GFP) in the epidermis, and (2) an mCherry-tagged plasma membrane marker, in ~1/3 of neurons (Pcnd-1 promoter; Figure 2A). To enhance the effectiveness of RNAi, both strains were also engineered to contain a pair of RNAi-sensitizing mutations (nre-1(hd20) lin-15b(hd126)20. These were constructed with the goal of performing a large-scale RNAi-based screen of the ~2,000 genes required for embryonic development. However, this pair of strains will also constitute a broadly useful standardized platform for assessing developmental defects in mutants, and for more detailed analyses of the roles of different proteins in development.
For data collection in this semi-high throughput format, with these strains, green, red and bright-field z-series (18 x 2 µm2 intervals) are collected, every ~20 min, for a period of 10 h in a 16 °C temperature-controlled room; this maintains the microscope between 21-23 °C during the run. The 20 min time interval allows us to image 50 fields of embryos (point visits) per experiment. Users can tailor acquisitions to shorter intervals with fewer point visits, or longer intervals with more point visits to best suit their experimental objectives. For these strains, the early developmental timepoints are not imaged because the tissue specific reporters driving marker expression do not begin to express until mid-to-late gastrulation. During this early phase, wells were scanned at low magnification (10x) and fields are selected for high magnification imaging. Selecting fields containing more than one early stage embryo is recommended, but avoiding fields where embryos are partially overlapping, overly crowded, or are found at the extreme edge of the well. Following overnight 60x imaging, a low magnification (10x) whole well scan is performed to determine whether embryos hatch with normal appearance (WT), hatch with visible abnormalities (larval abnormal), or are embryonic lethal for each condition assessed (Figure 1B). This step serves as an easy alternative to setting up plates in parallel to assess lethality across a larger population; the whole well 10x post-scan provides embryonic lethality data on 50-80 embryos per condition. Using this measure to compare the population embryonic lethality data for controls between runs is a useful control that ensures consistency with respect to the health of the strains and the environmental conditions and processing steps. When imaged in combination, the two custom reporter strains provide informative readouts for most major developmental events, including cell fate specification, dorsal intercalation, epidermal enclosure, elongation, and neurogenesis (representative control images are shown in Figure 2B, also see Wang et al.10).
Data processing: automated embryo cropping and orientation
With our imaging protocol, each high-resolution imaging field contains between 1 and 5 embryos; these embryos are randomly oriented and are frequently positioned immediately adjacent to one another. Data collected using conventional embryo mounting techniques suffers from the same challenges. To prepare the data for subsequent visualization and analysis, considerable hands-on manipulation is needed to crop and orient the embryo sequences, as well as to pre-process them to subtract background, correct for drift, and compensate for signal attenuation with depth. To streamline this process, a custom embryo cropping program was built that isolates and orients each embryo from the broader field (Figure 1C, Figure 3). This program has been packaged as a user-friendly, stand-alone program with graphical user interface (GUI, compatible with data from most imaging platforms) that can take in a 3D time-lapse sequence of a field of embryos, and process it into individual tiff series for each embryo in the field (Figure 3B, available at https://zenodo.org/record/3235681#.XPAnn4hKg2w)10,12. The Python source code for the GUI (embryoCropUI.py), and a batch version of this program (screenCrop.py) are also available for experienced Python users on GitHub (https://github.com/renatkh/embryo_crop.git)10,13. The core functionality of this program is to locate, crop, and orient the embryo along the anterior-posterior axis. Simultaneously, background subtraction, attenuation correction, and drift correction can be performed on the dataset using optional adjustable settings.
Briefly, the automated cropping software iteratively detects individual embryos in a binary mask obtained from the bright-field images and sequentially crops the embryos from all channels and orients the images along the anterior-posterior axis (Figure 3B, first described in Wang et al.10). Prior to cropping, the software corrects for drift, subtracts background and performs depth attenuation correction in each fluorescence channel (optional). To obtain the binary mask, the software applies Canny edge detection21 to the bright-field images, performs maximum intensity projection of the three central planes and fills in small holes. The algorithm detects individual embryos by fitting a Cassini oval to a section of the binary mask outline (see Figure 3B, 3C). After aligning the oval along its main axis, the software measures red fluorescence intensity in each half of the isolated embryo and rotates the embryo such that red intensity is oriented towards the left, which puts the embryo anterior on the left for both of the reporter strains used in this work (Figure 3D). Piloting of this software revealed that most embryos were efficiently cropped as anticipated. Minor imaging issues, such as embryo drift, temporary focal plane loss (due to bubbles in oil), or crowded fields of embryos did not typically disrupt successful cropping. However, for fields where there were large clumps of embryos (overlapping somewhat in z), embryos that were significantly tilted within the imaging plane (in Z), long-term focal plane loss, or embryos that were partially cutoff, successful cropping was not always achieved. These issues can be easily circumvented by better field selection during the data acquisition phase. Overall, pre-processing data collected with this semi-high-throughput method or using conventional mounting methods is a useful first step for visualizing and analyzing embryonic datasets.
Validation of image collection and data processing methods: visualize embryonic development after knock-down of 40 previously-described genes
To validate this semi-high-throughput image collection method and custom cropping regime, a small RNAi screen was performed in the custom strains, directed against 40 genes with previously described functions in cell fate specification and morphogenesis (described by Wang, Ochoa, Khaliullin and colleagues10,11). To do this, dsRNA was generated against each target, injected L4 animals from each strain, waited 20-24 h, and then dissected out and imaged embryos using the protocol described above (for complete dataset10,11). Achieving developmental phenotypes by RNAi requires inhibition of both maternal and zygotic gene expression. Developmental genes can be maternally expressed, with the resulting protein products being loaded into the embryos, or zygotically expressed in the embryo, or, in many cases, both10,22,23. The time between injection and embryo filming is the critical variable to effectively targeting maternally and zygotically expressed populations; timing determines both the amount of injected dsRNA that is loaded into the embryo to prevent zygotic expression24 and the extent of maternal protein depletion. After the dsRNA injection, the maternal mRNA corresponding to the targeted gene gets degraded and does not recover over in a relevant time interval. Pre-existing protein depletion requires embryo production, which ejects the maternal protein from germline tissues by loading it into forming embryos. 20-24 h at 20 °C is the shortest incubation time that allows consistent maternal depletion; maternal depletion gets better at later timepoints (i.e., 36-42 h after injection). The amount of injected dsRNA that is loaded into the embryo dictates how effective the prevention of zygotic gene expression will be. The amount of loaded RNA peaks about 5-10 h after injection, when embryos with injected material are first fertilized, declining thereafter. In our experience, 24 h after injection is an optimal timepoint, in which maternal protein depletion and inhibition of zygotic gene expression are both effective. Analysis of phenotypes in the custom reporter strains used in this work, across the set of 40 test genes, showed that our combined protocol yielded distinct, highly reproducible signature phenotypes across a broad spectrum of genes involved in cell fate specification and morphogenesis (see Wang et al.10); the complete dataset is available11). As an example of this data, still images of four different embryos for control, pha-4(RNAi), pal-1(RNAi), and mex-3(RNAi) in the germ layer reporter strain are shown in Figure 4A. For a more comprehensive discussion of the phenotypes observed in the 40-gene RNAi screen, see Wang et al.10
ImageJ macro: Compilation and visualization of all data for a given condition
Visualization of large numbers of embryos from individual tiff stacks can be tedious and time consuming to work through. From our initial effort, >400 individual embryos were isolated using the custom automated cropping program described above. To speed up first-pass analysis, we built an ImageJ macro, which generates an array of all embryos imaged for a given RNAi condition in a given strain background (Figure 4B). For each embryo in the array, a brightfield image and a maximum intensity projection image are presented side-by-side, for each time point, to generate a composite movie file. While this macro was written to function with our file structure, it can be edited to accommodate the user’s file structure. For best results, however, users should follow the naming and file structure conventions set forth in the protocol (see Zenodo or Github README files for more specifics for the Python and ImageJ application naming conventions). To view all of the ImageJ processed data and get more in-depth analysis of the phenotypes observed for the 40-gene test set, please see the data deposited in the Dryad database10,11. This ImageJ visualization format enables rapid assessment of the overall phenotype and its penetrance, and makes it easy to flag data quality issues, such as presence of debris or loss of focal plane. Overall, the combination of the semi-high-throughput data acquisition method, cropping program and ImageJ visualization tool, combined with custom reporter strains, greatly facilitates larger scale efforts to study embryonic development.

Figure 1. Overview of high-content imaging method, data processing procedure, and essential tools. (A) A comparison of the features of the standard agarose pad-based method for mounting C. elegans embryos to the semi-high-throughput multi-well imaging approach described here. (B) Overview of the semi-high-throughput method for monitoring embryonic development. See text for details. (C) Overview of the data processing and visualization tools, which can be used on data acquired using either a conventional agarose pad-based mounting procedure or the semi-high-throughput multi-well plate-based method described herein. A single 3 channel time lapse embryo sequence from one field (left) or multiple 3 channel, 4-dimensional fields of embryo data (right) can be processed using the custom cropping program, which is compatible with data captured on most imaging platforms. The graphical user interface (GUI) version of the program is user friendly and does not require any programming expertise. The screenCrop.py application requires Python expertise, but it has added capabilities- namely that it can crop in batch format. The output of this step is tightly cropped, anterior-posterior oriented embryo tiff stacks. To visualize all data from a single condition, an ImageJ macro was built that compiles cropped, rotated tiff stacks to show a brightfield image and maximum intensity projection for each embryo at each timepoint. (D) Panel shows essential tools needed for dissection and setup of semi-high-throughput imaging format. Some figure components reproduced with permission from Wang et al.10. Please click here to view a larger version of this figure.
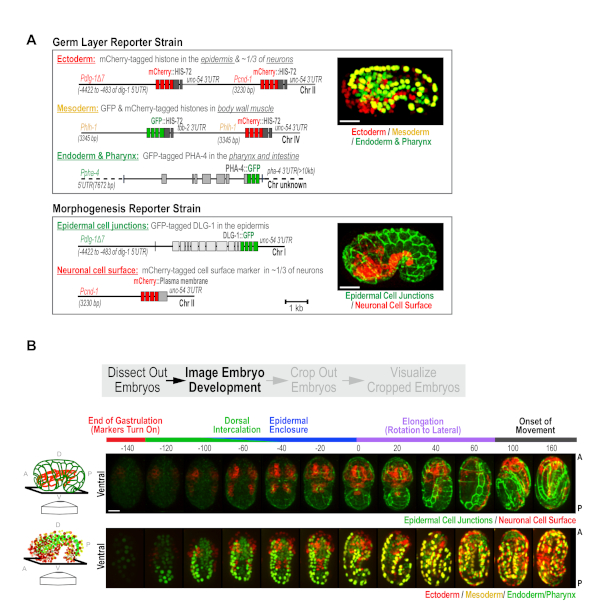
Figure 2. Custom strains generated for high-content imaging of C. elegans embryogenesis. (A) Schematics illustrate the transgenes used to construct the Germ Layer (top) and Morphogenesis (bottom) strains. (B) Maximum intensity projection panels show the developmental time-course in the Morphogenesis (top) and the Germ Layer (bottom) strains. Ventral view is shown, as illustrated in the schematics (left). Time is relative to the comma stage (t = 0), which is easily identifiable in both strains. Scale bar = 10 µm. Figure reproduced with permission from Wang et al.10. Please click here to view a larger version of this figure.
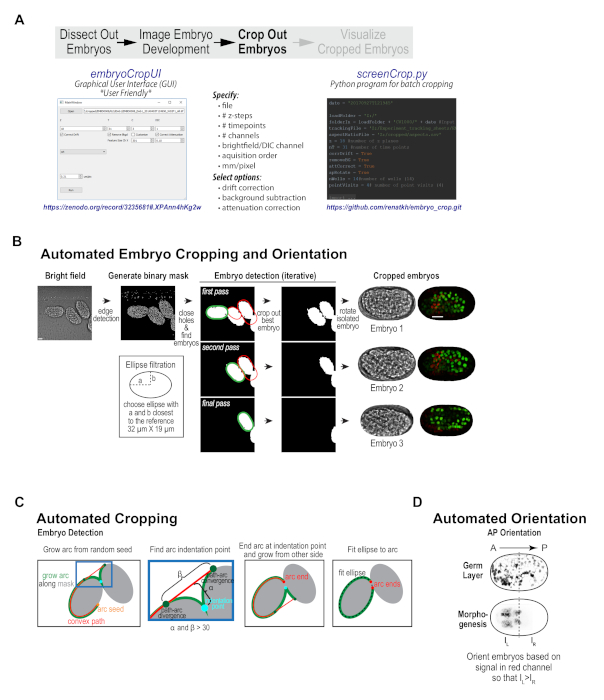
Figure 3. Custom cropping program isolates and orients individual embryos from imaging fields. (A) Schematic illustrates the features of the custom embryo cropping program and highlights the two options for accessing this program: a user-friendly GUI interface, which requires no programming expertise and is available on Zenodo (left) and a batch cropping version of the program, which requires Python experience and is available on Github (right). (B) Graphic summarizes the automated cropping algorithm — a binary mask is generated from 8-bit brightfield images and individual embryos are detected, cropped out, and oriented along the anterior-posterior axis. Scale bar is 10 µm. (C) Schematics detail the procedure used to iteratively detect embryos in the binary mask. (D) Schematic illustrates the method used for orienting embryos along the anterior-posterior axis. Panels B-D reproduced with permission from Wang et al.10. Please click here to view a larger version of this figure.

Figure 4. Custom ImageJ macro enables visualization of RNAi screening data by generating composite files for each condition. (A) Embryos for three example RNAi conditions from the 40-gene test set (see Wang et al.10,11 for complete dataset) are shown (right) that highlight the distinct signature phenotypes that are obtained for each condition and the reproducibility of the phenotypes within each condition. Panel was adapted with permission from Wang et al.10. (B) Panel illustrates how the custom ImageJ macro (OpenandCombine_embsV2.ijm) arrays embryos from a given RNAi condition into a composite ImageJ file that arrays the brightfield and maximum intensity projections for each embryo at each timepoint. While only one example is highlighted, this macro can compile the data for many RNAi conditions at once. ImageJ macro is available on Zenodo12. Scale bar = 10 µm. Please click here to view a larger version of this figure.
