Lysozyme was assayed with the stability screens and Protein X, a target protein in the Virus-X project, was assayed with the osmolyte screen. Both proteins generally produced melt curves with clearly-defined denaturation transitions in both TSA and nanoDSF experiments (see accompanying figures for representative curves). In a few cases where the samples that did not produce interpretable curves with a defined denaturation transition were interpreted as denatured and not included in Tm comparisons.
Figure 7 shows sample results from the salt screen, exemplifying the thermally-stabilising properties of ammonium chloride towards lysozyme. Concentration dependencies such as that shown above are often indicative of promising conditions, but it can be useful to compare the results of increasing ion concentration with several different salts to see if thermal stabilisation generally arises from an increase in buffer ionic strength or if the presence of specific ions confers additional stability.
Comparison of Tm values of lysozyme with the pH screen (Figure 8) reveals two pieces of information. Firstly, there is a general trend of increasing stability with decreasing pH values. Secondly, the range of Tm values obtained using different buffer systems with identical pH values can be significant.
Data in Figure 8 suggests that the agreement between TSA and nanoDSF in this experiment is generally good, but nanoDSF shows a tendency to identify slightly higher Tm values and slightly larger Tm shifts than TSA. However, some wells with pH values above 8.5 show large discrepancies between Tm values obtained from TSA and nanoDSF. Differences could potentially be attributed to the denaturation mechanism of the protein at different pH values; for example, a hydrophobicity-sensitive dye could give a comparatively low Tm reading if hydrophobic regions of a protein become exposed to solvent significantly faster than the environment of tryptophan residues changes in polarity.
Figure 9 shows a heatmap of Tm values obtained with lysozyme using the osmolyte screen. Conditions with the highest Tm increases compared to the control wells (wells A1 and A2, containing deionized water) are colored dark blue. Especially stabilizing conditions identified in Figure 9 include glycerol, 1 M D-sorbitol, 100 mM hypotaurine and 10 mM Ala-Gly (wells A4-A6, A9, E7 and F8, respectively).
Figure 10 shows a heatmap of Tm values obtained with Protein X. TSA and nanoDSF experiments with the Osmolyte screen reveal that the majority of osmolytes tested give either a minor increase in Tm (within 1 °C) or have a detrimental effect on Protein X's stability. In particular, dipicolinic acid at a concentration of 10 mM (well D1) appears to denature the sample at room temperature. The TSA and nanoDSF results quickly identify dipicolinic acid as an incompatible additive for Protein X which should be avoided when working with the protein. Nevertheless, high concentrations of D-sorbitol and arabinose (wells A9 and B9, both at 1 M) as well as glycerol and TMAO (wells A4-A6 and E1, respectively) were identified as thermally-stabilizing.
For lysozyme, combinations of conditions yielding the highest Tm values from each stability screen were tested to probe for a synergistic combined effect. Figure 11 shows a general increase in Tm values as more components of the buffer system (pH, salt and osmolyte) are added. In the case of MES, ammonium sulphate and D-sorbitol, a Tm increase as large as 10 °C can be observed when all components are present compared to MES alone. Figure 11 shows that a noticeable synergistic effect can occur when individual components of a buffer are optimized and combined with the stability screens.
On a more general note, Figure 11 also illustrates the magnitude of ΔTm values that can be observed in TSA and nanoDSF experiments. The magnitude of ΔTm achievable varies significantly based on the protein system, but any ΔTm value around and above 5 °C is often indicative of a beneficial stabilizing effect.

Figure 1: Bioprospecting. Sample collection from an extreme environment in the Virus-X Project. Please click here to view a larger version of this figure.
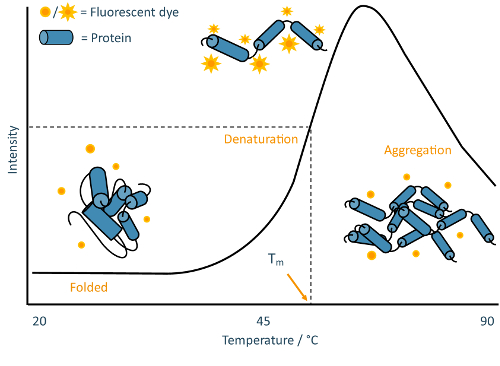
Figure 2: TSA schematic. Annotated example of a typical melt curve obtained from a TSA experiment. This curve is characteristic of classic "two-state" protein unfolding, where the sample population transitions from folded to denatured without detectable partially-folded intermediates. Please click here to view a larger version of this figure.
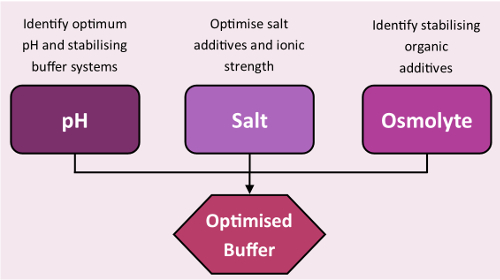
Figure 3: Stability screens workflow. Standard workflow of buffer optimization using the stability screens. Please click here to view a larger version of this figure.

Figure 4: Workflow of a standard TSA experiment with the stability screens. From left to right: (A) Pipetting aliquots of the stability screens into a 96-well plate. (B) Pipetting protein sample with fluorescent dye into the plate. (C) Sealing the plate before centrifugation. (D) Placing the plate into an RT-PCR system. Please click here to view a larger version of this figure.

Figure 5: Workflow of a standard nanoDSF experiment. (A) Opening the capillary loading rack. (B) Loading the capillaries into the rack. (C) Immobilizing the capillaries with the magnetic sealing strip. (D) Programming an experiment. Please click here to view a larger version of this figure.
Figure 6: User interface for the RT-PCR system. A TSA experiment has been programmed. Please click here to view a larger version of this figure.
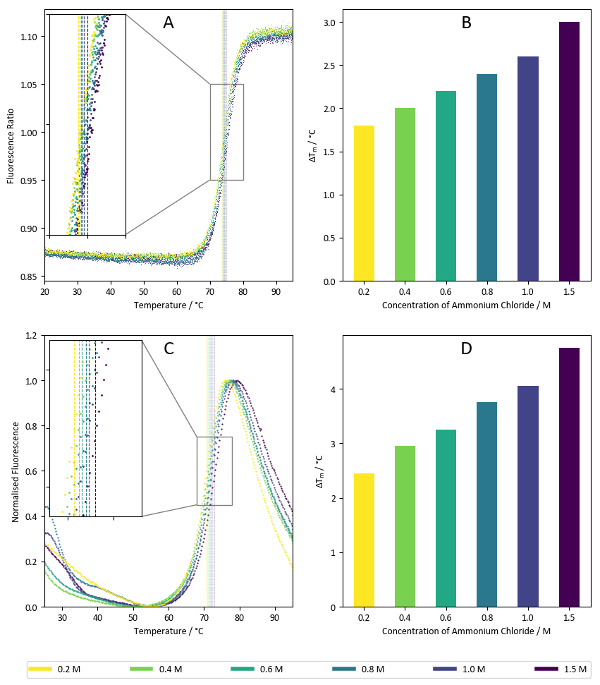
Figure 7: Salt screen sample data. (A) Ratio of fluorescence intensities at 350 nm vs 330 nm for a label-free nanoDSF experiment with lysozyme. Samples correspond to the wells C7-C12 of the salt screen (1.5 M – 0.2 M ammonium chloride). Calculated Tm values for each condition are superimposed on the plot. (B) Summary of Tm values calculated from the data presented in Figure 7A. (C) Fluorescence intensities at 590 nm for a TSA experiment using lysozyme with a hydrophobicity-sensitive reporter dye. Like Figure 7A, the samples correspond to the wells C7-C12 of the salt screen. Calculated Tm values are superimposed on the graph. (D) Summary of Tm values calculated from data present in Figure 7C. Please click here to view a larger version of this figure.
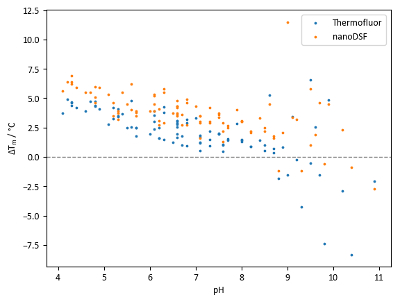
Figure 8: pH screen sample data. Summary of Tm shifts obtained with lysozyme and the pH screen. Each point represents an independent condition; points at the same pH value are of different buffer systems at the same pH. Tm shifts are calculated relative to a control Tm of 68.0 °C for TSA experiments and 71.9 °C for nanoDSF experiments. Please click here to view a larger version of this figure.
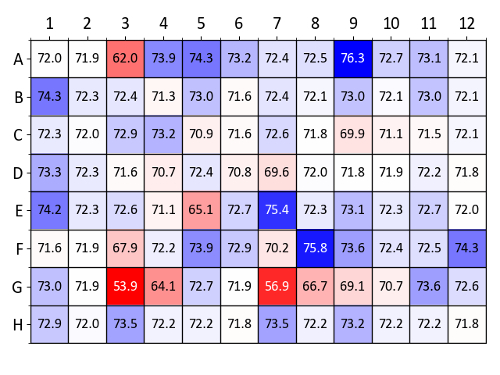
Figure 9: Osmolyte screen sample data (lysozyme). Summary of Tm values obtained from a label-free nanoDSF experiment with lysozyme and each well of the osmolyte screen. Tm values (in °C) are compared to the wells A1 and A2 which contain water as a control. A heatmap was generated based on ΔTm values compared (blue denotes a Tm increase and red a Tm decrease). Please click here to view a larger version of this figure.
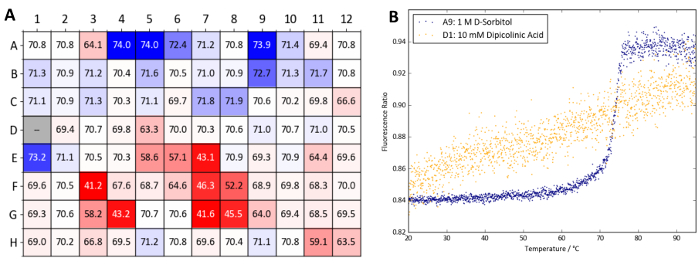
Figure 10: Osmolyte screen sample data (Protein X). (A) Summary of Tm values obtained from a label-free nanoDSF experiment with Protein X and the osmolyte screen. Tm values are compared to wells A1 and A2 which contain water as a control. A heatmap was generated based on ΔTm values compared (blue denotes a Tm increase and red a Tm decrease). (B) nanoDSF curves obtained from well A9 (1 M D-sorbitol, a stabilizing condition) and D1 (10 mM dipicolinic acid, a destabilizing condition). Please click here to view a larger version of this figure.
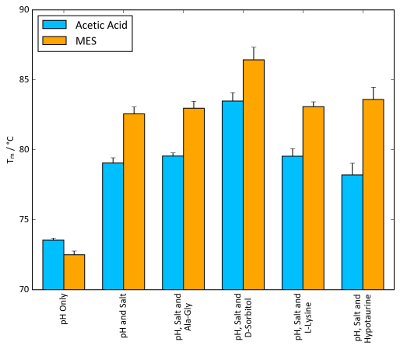
Figure 11: Buffer optimization effect on Tm. TSA Tm values of lysozyme combined with the conditions of each screen that afforded the largest increase in Tm. 100 mM acetic acid, pH 4.2, and 100 mM MES, pH 5.6, were chosen as the buffer systems, alongside 1.5 M ammonium sulphate as the salt. Osmolyte concentrations were identical to those found in the osmolyte screen: 10 mM Ala-Gly, 1 M D-sorbitol, 50 mM L-lysine and 100 mM hypotaurine. Error bars represent the standard deviation of six replicates. Please click here to view a larger version of this figure.
| Molecule | PDB Code | Frequency of Co-crystallisation |
| Phosphate | PO4 | 5132 |
| Acetate | ACT | 4521 |
| 2-(N-Morpholino)-Ethanesulfonic Acid (MES) | MES | 1334 |
| Tris(hydroxymethyl)aminomethane (tris) | TRS | 1155 |
| Formate | FMT | 1072 |
Table 1: Summary of buffer molecule co-crystallization frequency in Protein Data Bank (PDB) entries. Data obtained through PDBsum16 from a total of 144,868 entries (correct as of 12-5-18).
Supplementary Information. Please click here to download this file.
Supplementary Table 1. Please click here to download this file.
Supplementary Table 2. Please click here to download this file.
Supplementary Table 3. Please click here to download this file.
Critical aspects within the protocol include the centrifugation step and proper sealing of the 96-well plate for TSA experiments (step 1.5). Centrifugation ensures that the protein sample and screen condition come into contact and mix. Additionally, if an unsealed plate is used for a TSA experiment, there is a significant risk of solvent evaporating throughout the experiment, causing an increase in sample concentration and increasing the chance of premature protein aggregation.
TSAs and nanoDSF are amenable to a wide range of protein samples; the vast majority of samples can produce interpretable melt curves with a hydrophobicity-based reporter dye or through dye-free nanoDSF. If standard fluorescence sources are not suitable for your protein, the simplest modification to the protocol that could be explored is the choice of fluorophore. Several alternative dyes could be suitable for TSA experiments. Examples include N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM), a compound that fluoresces after reacting with a thiol27, and 4-(dicyanovinyl)julolidine (DCVJ), a compound that varies its fluorescence based on the rigidity of its environment, increasing its fluorescence as a protein sample unfolds28,29 (the latter dye often requires high concentrations of sample).
Alternative methods of melt curve analysis are available if Tm is not automatically calculated by the instrument software. If data is homogenous and only one denaturation step is apparent in the melt curves, a truncated dataset can be fitted to a Boltzmann sigmoid with the following equation:
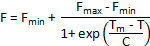
Where F is the fluorescence intensity at temperature T, Fmin and Fmax are the fluorescence intensities before and after the denaturation transition, respectively, Tm is the midpoint temperature of the denaturation transition and C is the slope at Tm. While this method works well for simple two-step denaturation processes, it is unsuitable for complex melt curves with multiple transitions.
One of the major advantages of TSA is its accessibility; TSA experiments can be performed in any RT-PCR system with filters at suitable wavelengths for the fluorescence dye employed. This coupled with the low cost of consumables, ease of operation and relatively low amount of protein needed, make TSA a valuable technique for a wide range of project scales, both in industry and academia.
As well as indicating favorable buffer conditions, the screens contain some wells that may give clues to the presence of structural metals within a sample protein. Wells that may be of particular interest in the salt screen are G6 and G7, which contain 5 mM EDTA and 5 mM EGTA, respectively. Significant thermal destabilization in these wells may be indicative of important metal ions in the protein that are sequestered by the chelants. Compounds within the osmolyte screen can also potentially provide clues to the function of a protein. Many of the compounds in the screen belong to classes of molecule that are common substrates of enzymes. For example, the general stabilization afforded by saccharides (present in wells A11-B10) for lysozyme could be attributed to their structural similarity to established substrates of the enzyme, N-acetylglucosamine oligomers30.
The TSA and nanoDSF protocols outlined above can also be adapted to study protein-ligand interactions. Ligands that bind specifically to a protein can increase its thermal stability by introducing new interactions within the complex. A dose-dependent positive shift in protein Tm is a promising sign of a successful protein-ligand interaction. The speed, throughput and low cost of screening compound libraries with TSAs has made it a very popular method in early-stage drug discovery.
Optimizing the buffer conditions of target proteins and their ligand complexes can be essential for a project's success, as many literature examples demonstrate31,32,33,34. With a typical assay taking under 2 h including setup time, TSAs and nanoDSF coupled with stability screens represent a fast, inexpensive technique for buffer optimizations.
