Preparation of Mouse Vomeronasal Organ for Vibratome Sectioning
Abstract
Source: Ackels, T., et al. In-depth physiological analysis of defined cell populations in acute tissue slices of the mouse vomeronasal organ. J. Vis. Exp. (2016)
This video outlines the isolation of a mouse vomeronasal organ (VNO) and tissue preparation for vibratome sectioning. Sections are used to characterize the biophysical and physiological attributes of a specific group of sensory neurons.
Protocol
All procedures involving animal models have been reviewed by the local institutional animal care committee and the JoVE veterinary review board.
Both C57BL/6 mice and Fpr-rs3-i-Venus mice were housed in groups of both sexes at room temperature on a 12-hour light/dark cycle with food and water available ad libitum. For experiments, young adults (6-20 weeks) of either sex were used. No obvious gender-dependent differences were observed.
1. Solution Preparation
- Prepare extracellular solution S1: 4-(2-Hydroxy-ethyl)piperazine-1-ethanesulfonic acid (HEPES) buffered extracellular solution containing (in mM) 145 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES; pH = 7.3 (adjusted with NaOH); osmolarity = 300 mOsm (adjusted with glucose).
- Prepare extracellular solution S2: Carbogen-oxygenated (95% O2, 5% CO2) extracellular solution containing (in mM) 125 NaCl, 25 NaHCO3, 5 KCl, 1 CaCl2, 1 MgSO4, 5 BES (Bis(2-hydroxyethyl)-2-aminoethanesulfonic acid); pH = 7.3; osmolarity = 300 mOsm (adjusted with glucose).
- Prepare 4% low-gelling temperature agarose solution (S3) for tissue embedding: 4% low-gelling temperature agarose. Dissolve 2 g low-gelling agarose in 50 ml of S1 in a 100 ml glass laboratory bottle together with a magnetic stirrer. To melt the agarose, put the bottle in a water bath (with the lid not tightly closed) at 70 °C for approximately 20 min or until the solution becomes transparent while stirring. Cool down and keep melted agarose in a second water bath at 42 °C until further use.
- Prepare intracellular solution S4: Pipette solution containing (in mM) 143 KCl, 2 KOH, 1 EGTA (ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid), 0.3 CaCl2 (free Ca2+ = 110 nM), 10 HEPES, 2 MgATP(Magnesium Adenosine 5'-triphosphate), 1 NaGTP(Guanosine 5′-triphosphate sodium salt hydrate); pH = 7.1 (adjusted with KOH); osmolarity = 290 mOsm.
- Modify/adjust the composition of S1, S2, and S4 according to individual experimental design (e.g., use pharmacological blockers to isolate certain types of voltage-activated currents).
2. Workspace Preparation
- Fill the oxygenating slice storing chamber with S2 at least 30 min prior to dissection, place the slice on ice for temperature and pH adjustment, and oxygenate the solution continuously.
- Fill the reservoir for slice superfusion at recording setup with S2 and oxygenate continuously.
- Prior to dissection, fill the vibratome chamber with S2 and arrange crushed ice around the chamber. Alternatively, transfer the spare ice-cold oxygenated solution from the oxygenating chamber into the vibratome chamber and keep oxygenating continuously.
- Arrange surgical tools and consumables.
- Place the ice gel pack under the dissecting microscope and cover it with a paper towel to prevent the VNO tissue from freezing to the bottom of the dish.
- Clean the razor blade by briefly rinsing it in 70% ethanol and distilled water, then mounting it to the vibratome. Replace it for every slicing session.
3. VNO Dissection and Embedding
- Sacrifice the animal by brief exposure to CO2 and decapitate it using sharp surgical scissors. Note: As time from sacrificing the animal to putting the VNO capsule on ice is critical, minimize the time to less than 2 min. To maintain tissue viability, embed both fully dissected VNOs in agarose in less than 30 min.
- Remove the lower jaw with large surgical scissors. Enter through the mouth cavity and cut the mandible bones and muscles of each side separately.
- Place the remaining part of the head upside down in the large Petri dish.
- Pull away the skin of the upper jaw and around the tip of the nose with medium forceps to gain better access to the incisors.
- Use bone scissors to cut away the largest part of the incisors at a ~45° angle in the rostral direction (Figure 1A). This will ease the removal of the VNO capsule from the nasal cavity.
Note: Do not cut to the root of the tooth to prevent damage to the tip of the VNO capsule. - Grab the rigid upper palate at its rostral part with medium forceps and carefully peel back in one piece at a flat angle (Figure 1B).
Note: Repeatedly rinse with ice-cold S2. - Use microspring scissors to cut the bony fusion between the tip of the vomer bone and the jaw. Insert the scissor tips with the curved part of the tip pointing outward away from the VNO and carefully cut the bone in small steps on both the left and right sides lateral to the VNO capsule.
- To remove the VNO capsule, use micro spring scissors to cut through the vomer bone at the caudal part and carefully lift it out of the nasal cavity using medium forceps. Immediately transfer the VNO to a small petri dish under a stereo microscope on an ice gel pack, where the remaining steps of the dissection will be performed.
- Rinse the VNO in a small amount of ice-cold S2 to prevent the tissue from drying out.
- Separate the cartilaginous capsules that contain the VNO soft tissues by grabbing the back of the vomer bone with medium forceps. Position the capsule for a dorsal view so that a split between both VNOs becomes visible (Figure 1Di).
- Use the tip of fine forceps to separate both cartilage VNO capsules from the central bone while keeping the vomer bone pinned down at the rear part.
Note: Use forceps only at the rim of the cartilage capsule, and be very careful not to pierce through the cartilage as the delicate sensory epithelium is easily damaged. - Once the two VNOs are separated, start removing the cartilage encapsulating the first VNO.
- Grab the top rim of the capsule with one fine forceps and split away the cartilage wall that was previously attached to the vomer bone (medial side).
- To remove the remaining cartilage, turn the VNO with its curved lateral side to the bottom of the dish and securely pin down the cartilage on one side using forceps. Carefully move the second fine forceps from the back side at a very flat angle between the cartilage and VNO to loosen the connection between tissue and cartilage.
- To avoid damaging the sensory epithelium, slowly peel the VNO away from the cartilage by holding it at its caudal tip.
- Once the VNO is levered from the capsule, remove all remaining small cartilage parts. Any remaining pieces of cartilage will detach the tissue from the surrounding agarose during the slicing process.
- Place a small drop of ice-cold S2 on the first dissected VNO to prevent tissue damage. A large blood vessel on the lateral side will become visible, indicating that the organ is still intact and was not grossly damaged during the dissection. In case the blood vessel got damaged, it will still be worthwhile to carry on with slicing the VNO as long as the overall morphology was not clearly impaired.
- Immediately dissect the second VNO.
- To embed the VNOs, fill both small Petri dishes to the rim with melted S3 (stored in a water bath at 42 °C; see 1.3).
- Hold the VNO on the broader caudal end with fine forceps to avoid damage to the sensory epithelium.
- Immerse the VNO in the agarose and move it horizontally back and forth several times to remove the film of the extracellular solution as well as any air bubbles from its surface.
- Position the VNO vertically, with the caudal tip facing the bottom of the dish. Instead of directly pinching the VNO with the forceps, move the forceps tip in close proximity to the VNO to adjust orientation.
Note: Orientation during embedding is crucial as it determines slice plane and accessibility to sensory neurons during the experiment. - Place dishes on a gel ice pack and wait until the agarose has solidified.
Note: Do not change VNO orientation once the agarose solidifies, as this will detach the tissue from the surrounding agarose.
4. Coronal VNO Tissue Slicing
- Use a small spatula to remove the agarose block from the small dish into the lid of a large Petri dish. Flip the agarose upside down, leaving the caudal tip of the VNO facing upwards.
- Cut the block into a pyramidal shape using a surgical scalpel (3-4 mm at the tip, 8-10 mm at the bottom). Take care not to damage the embedded tissue.
- Use super glue to fix the pyramid-shaped block to the center of the vibratome specimen plate and wait ~1 min for the glue to dry completely.
- Transfer the specimen plate to the slicing chamber and prepare the second VNO accordingly. Keep a plate with the second specimen at 4 °C until use.
- Use a vibrating blade microtome with the following settings: thickness: 150-200 µm; speed: 3.5 a.u. = 0.15 mm/sec; frequency: 7.5 a.u. = 75 Hz. Transfer slices to an oxygenating chamber until use after briefly inspecting slice morphology under the dissecting microscope. Slices can be kept for several hours.
Representative Results
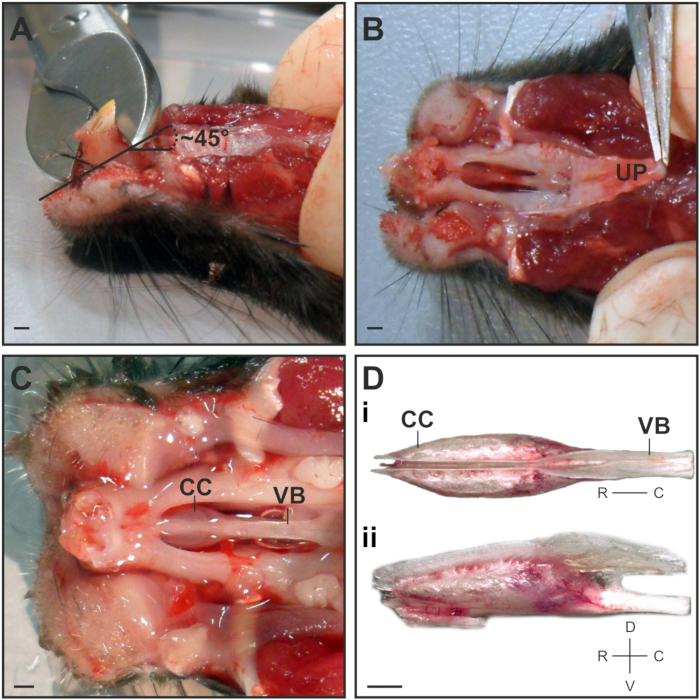
Figure 1: Dissection of the VNO. (A) Side view on the mouse head to illustrate the position and angle at which the incisors are cut. (B) Ventral view depicting the best point to grab and peel back the upper palate (UP). (C) Ventral view onto the cartilage capsule (CC) that harbors the VNO and the vomer bone (VB) after removing the lower jaw, incisors and palate. (D) Dorsal view of the dissected VNO capsule depicting the bilateral localization of both VNOs (Di). The lateral view illustrates the rim of cartilage on the dorsal side where both VNOs need to be separated (Dii). Scale bar = 1 mm (A–D).
Disclosures
The authors have nothing to disclose.
Materials
| Agarose (low-gelling temperature) | PeqLab | 35-2030 | |
| ATP (Mg-ATP) | Sigma-Aldrich | A9187 | |
| Bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES) | Sigma-Aldrich | B9879 | |
| Calcium chloride | Sigma-Aldrich | C1016 | |
| Ethylene glycol tetraacetic acid (EGTA) | Sigma-Aldrich | E3889 | |
| Glucose | Sigma-Aldrich | G8270 | |
| GTP (Na-GTP) | Sigma-Aldrich | 51120 | |
| (2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) | Sigma-Aldrich | H3375 | |
| Magnesium chloride | Sigma-Aldrich | M8266 | |
| Potassium chloride | Sigma-Aldrich | P9333 | |
| Potassium hydroxide | Sigma-Aldrich | 3564 | |
| Sodium chloride | Sigma-Aldrich | S7653 | |
| Sodium hydrogen carbonate | Sigma-Aldrich | S5761 | |
| Sodium hydroxide | Sigma-Aldrich | S8045 | |
| Large petri dish, 90 mm | VWR | decapitation, dissection of VNO capsule | |
| Small petri dish, 35 mm | VWR | lid for VNO dissection, dish for embedding in agarose | |
| Sharp large surgical scissor | Fine Science Tools | decapitation, removal of lower jaw | |
| Strong bone scissors | Fine Science Tools | cutting incisors | |
| Medium forceps, Dumont tweezers #2 | Fine Science Tools | removing skin and palate | |
| Micro spring scissors, 8.5 cm, curved, 7 mm blades | Fine Science Tools | cutting out VNO | |
| Two pairs of fine forceps, Dumont tweezers #5 | Fine Science Tools | dissecting VNO out of cartilaginous capsule | |
| Small stainless steel spatula | Fine Science Tools | handling agarose block and tissue slices | |
| Hot plate magnetic stirrer | Snijders | 34532 | |
| Microforge | Narishige | MF-830 | |
| pH Meter five easy | Mettler Toledo | ||
| Pipette storage jar | World Precision Instruments | e212 | |
| Razor blades | Wilkinson Sword GmbH | Wilkinson Sword Classic | |
| Oxygenating slice storage chamber; alternative commercial chambers are e.g. BSK1 Brain Slice Keeper (Digitimer) or the Pre-chamber (BSC-PC; Warner Instruments) | custom-made | ||
| Stereo microscope | Leica Microsystems | S4E | |
| Vibratome | Leica Microsystems | VT 1000 S | |
| Water bath | Memmert | WNB 45 |
