1. RNA dual end-labeling
NOTE: The following protocol describes the site-specific labeling of RNAs with a FRET pair of fluorophores by covalent attachment of a donor dye (sCy3) to the 5'-phosphate and an acceptor dye (Cy5) to the 3'-ribose. A catalytically active long RNA, the group II intron ribozyme, is chosen as the RNA of interest. Table 1 and Figure 1 summarize this dual-end labeling protocol. Perform all steps involving fluorophores under dark conditions.
| Day 0 | ▪ Aliquot 50-75 µg of RNA to a total volume of 55 µL per 1.5 mL tube. | |||||
| Day 1 | 5′-Phosphate activation | |||||
| ▪ Add 45 µL of freshly prepared EDC-NHS, pH 6.0 solution to the RNA in ddH2O to a final volume of 100 μL, mix well and incubate for 4 h at 25 °C and 500 rpm. | ||||||
| ▪ Purification round 1: Overnight EtOH precipitation. | ||||||
| Day 2 | ▪ Precipitate the 5′-activated RNA, wash, and dry. | |||||
| 5′-Dye attachment | ||||||
| ▪ Resuspend in 95 µL of 100 mM MOPS, pH 7.5. | ||||||
| ▪ Add 5 μL of 2 mM amine-functionalized dye solution. | ||||||
| ▪ Mix well and incubate for 16 h at 25 °C and 500 rpm. | ||||||
| Day 3 | ▪ Purification round 2: EtOH precipitation. | |||||
| Day 4 | ▪ Precipitate the 5′-activated RNA, wash, and dry. | |||||
| Blocking step | ||||||
| ▪ Resuspend in 100 μL of 100 mM Tris–HCl, pH 7.5, and incubate for 2 h at 25 °C and 500 rpm. | ||||||
| ▪ Purification round 3: Centrifugal filtration. | ||||||
| → Elute the 5′-labeled RNA. | ||||||
| Day 5 | 3′-Periodate oxidation | |||||
| ▪ Incubate the RNA with 20 mM NaIO4 in 50 mM NaOAc buffer, pH 5.5 in a final volume of 100 μL for 2 h at 25 °C and 500 rpm. | ||||||
| ▪ Quench the excess periodate: Add 30 μL of 50% glycerol, mix well, and incubate for 30 min at 25 °C and 500 rpm. | ||||||
| ▪ Purification round 4: Overnight EtOH precipitation. | ||||||
| Day 6 | ▪ Precipitate the 3′-oxidized RNA, wash, and dry. | |||||
| 3′-Dye attachment | ||||||
| ▪ Resuspend in 95 µL of 50 mM NaOAc, pH 6.0. | ||||||
| ▪ Add 5 μL of 2 mM hydrazide-functionalized dye solution. Mix well and incubate for 16 h at 25 °C and 500 rpm. | ||||||
| Day 7 | ▪ Purification round 5: EtOH precipitation. | |||||
| Day 8 | ▪ Precipitate the labeled RNA, wash, and dry. | |||||
| ▪ Centrifugal filtration. | ||||||
| → Elute the dual-end labeled RNA. | ||||||
Table 1: Protocol summary for RNA dual-end labeling. Please click here to download this Table.
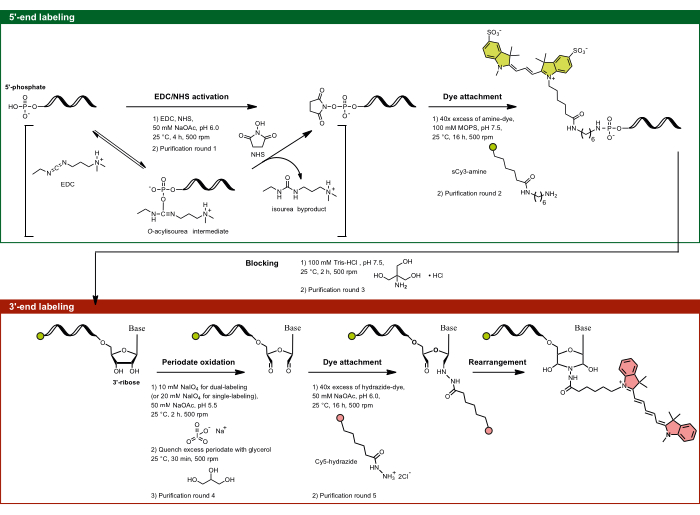
Figure 1: The experimental flow of dual-end labeling by targeting the 5'-phosphate and the 3'-ribose sugar. The 5'-phosphate is activated using EDC in the presence of NHS and is subsequently coupled with the amine-functionalized dye. The 3'-diol moiety of RNA is oxidized by periodate activity to dialdehyde, which further reacts with the hydrazide-functionalized dye. For dual-end labeling, it is important to start with the 5'-labeling to prevent cross-labeling, followed by 3'-labeling with an intermediate blocking step. Abbreviations: EDC = carbodiimide; NHS = N-hydroxysuccinimide; MOPS = 3-morpholinopropane-1-sulfonic acid; NaOAc = sodium acetate. Please click here to view a larger version of this figure.
- 5'-end labeling: Targeting the 5'-phosphate by EDC/NHS activation
NOTE: This 5'-labeling method applies not only to RNA but to any single-stranded nucleic acid containing a 5'-phosphate. The specificity of the reaction is governed by pH dependence, allowing only the 5'-phosphate to be site-specifically targeted, despite the presence of multiple phosphates in the RNA backbone. At pH 6.0, the 5'-phosphate's unique reactivity toward carbodiimides is due to its specific protonation state, where two oxygens are deprotonated, and one remains protonated. This makes the 5'-phosphate reactive, while the fully deprotonated backbone phosphates remain unreactive, enabling selective labeling of the 5'-end via EDC/NHS targeting.- Prepare the EDC-NHS-NaOAc, pH 6.0 solution. Mix 1.5 mg of EDC and 2.0 mg of NHS per aliquot in 35 µL of ddH2O and 10 µL of 0.5 M NaOAc, pH 6.0 (pH adjusted with glacial acetic acid).
- Aliquot the RNA of interest to have approximately 50-75 µg RNA per tube in 55 µL of ddH2O. Add 45 µL of the EDC-NHS-NaOAc, pH 6.0 mixture to the RNA, reaching a total volume of 100 µL with the final concentrations of 78 mM EDC, 174 mM NHS, and 50 mM NaOAc, pH 6.0. Incubate for 4 h at 25 °C while shaking at 500 rpm.
NOTE: Here, 70 µg corresponds to 250 pmol of an in vitro transcribed 915 nt RNA and 2.5 µM in the final reaction volume of 100 µL. - Purification round 1: Purify the 5'-phosphate-activated RNA by EtOH precipitation.
- Prepare a 2 mM sulfonated Cyanine3 amine (sCy3-amine) solution in water.
- Resuspend the activated RNA pellet in 95 µL of 100 mM 3-morpholinopropane-1-sulfonic acid (MOPS) buffer, pH 7.5 (pH adjusted with NaOH). Add 5 µL of 2 mM sCy3-amine solution to the activated RNA. Couple the fluorophore to the activated 5'-phosphate by incubation for 16 h at 25 °C, while shaking at 500 rpm.
NOTE: Store MOPS buffer at 4 °C in the dark. NaOH is chosen for pH adjustment to keep the group II intron ribozyme inactive. The fluorophore should be at least 40-fold excess of the RNA. - Purification round 2: Increase the volume by adding 200 µL of ddH2O to improve the separation. Purify the 5'-phosphate-labeled RNA by EtOH precipitation and repeat until the supernatant is colorless (usually two rounds are needed).
- Blocking step: Resuspend the RNA in 100 µL of 100 mM Tris-HCl, pH 7.5, and incubate for 2 h at 25 °C and 500 rpm. For 5'-end single-labeling only, skip the blocking step, resuspend the RNA in ddH2O, and proceed with step 1.1.8. However, for simplicity, if dual-labeling, separate the 5'end single-labeled control only after step 1.1.9.
NOTE: This step serves to block the activated 5'-phosphates that have not been coupled to an amine-functionalized fluorophore by reacting with a relatively smaller primary amine source (e.g., Tris) to minimize the risk of cross-labeling with the hydrazide-functionalized fluorophore that is used for the 3'-end labeling protocol. - Purification round 3: Remove the free dyes by washing the labeled RNA by centrifugal filtration with a total of at least 10 mL of ddH2O, then elute in ddH2O.
NOTE: The molecular cut-off of the filter should be less than half the size of the nucleic acid. Buffer of choice can be used instead of ddH2O. Elution should follow the manufacturer's instructions, ensuring the sample is not spun to complete dryness. - Determine the RNA and conjugate dye concentrations by UV-Vis spectroscopy.
- 3'-end labeling: Targeting the 3'-ribose by periodate oxidation
- Aliquot the 5'-end labeled RNA in ddH2O to have approximately 50-75 µg of RNA per tube in 90 µL. If the elution volume of the previous step was high, resulting in a low concentration, concentrate the RNA by precipitating and resuspending the pellet in ddH2O. Add 5 µL of 1.0 M NaOAc buffer, pH 5.5 (corresponding to 50 mM NaOAc, pH 5.5 for 100 µL reaction volume).
NOTE: Similar to the 5'-end single-labeled RNA, we routinely prepare 3'-end single-labeled RNA as a control. To do this, aliquot the unlabeled RNA to have approximately 50-75 µg of RNA per tube in 86 µL of ddH2O. - Add 4 µL of freshly prepared 500 mM sodium meta-periodate (NaIO4) stock solution (corresponding to 10 mM NaIO4 for 100 µL reaction volume). For 3'-end single labeling, add 8 µL of NaIO4 stock solution (corresponding to 20 mM NaIO4 for 100 µL reaction volume). Mix thoroughly and incubate for 2 h at 25 °C while shaking at 500 rpm under dark conditions as NaIO4 is light-sensitive.
NOTE: Do not change the order of addition or further increase the incubation time, as this may cause photobleaching of the attached donor dye. The concentration of NaIO4 used is decreased by half for dual-end labeling to minimize the quenching of the already attached dye at the 5'-end. - Stop the reaction by adding 30 µL of 50% glycerol. Incubate for 30 min at 25 °C while shaking at 500 rpm.
NOTE: Glycerol serves as a diol to quench the excess of periodate. - Purification round 4: Add 400 µL of ice-cold EtOH-NaOAc precipitation mixture and perform ethanol precipitation.
- Resuspend the oxidized RNA pellet in 95 µL of 50 mM NaOAc, pH 6.0.
- Hydrazide-dye attachment: Prepare the fluorophore solution by dissolving a few crystals of Cyanine5 hydrazide (Cy5-hydrazide) in DMSO and then diluting with ddH2O to the concentration of 2 mM. If the fluorophore of choice is water-soluble, prepare the solution in ddH2O. Add 5 µL of 2 mM Cy5-hydrazide solution to the oxidized RNA. Mix well and incubate for 16 h at 25 °C while shaking at 500 rpm.
NOTE: The fluorophore should be at least 40-fold excess of the RNA. - Purification round 5: Purify the dual-end-labeled RNA (or 3'-end single labeled RNA) by EtOH precipitation and centrifugal filtration, similar to steps 1.2.6 and 1.2.8, respectively, and elute in ddH2O.
- Calculate the RNA and conjugate dye concentrations by UV-Vis spectroscopy.
- Characterize the labeled RNA using analytical gel-based assays, ensemble fluorescence spectroscopy (see the Representative Results section), and/or analytical HPLC as shown elsewhere3.
- Aliquot the 5'-end labeled RNA in ddH2O to have approximately 50-75 µg of RNA per tube in 90 µL. If the elution volume of the previous step was high, resulting in a low concentration, concentrate the RNA by precipitating and resuspending the pellet in ddH2O. Add 5 µL of 1.0 M NaOAc buffer, pH 5.5 (corresponding to 50 mM NaOAc, pH 5.5 for 100 µL reaction volume).
2. Microfluidic chamber preparation
NOTE: We recommend handling six or eight chambers at a time. Sonication is performed at room temperature unless otherwise indicated. This protocol limits the use of organic solvents, such as acetone, to prevent the solubilization of trace impurities. For alternatives, refer to references15,16.
- Cleaning
- Drill four holes in the quartz slides with a diamond driller according to the scheme given in Figure 2A to form two channels.
NOTE: Although they may break more easily during drilling, glass slides are a cost-effective alternative to quartz slides for objective-based TIRF microscopy setups where the coverslip is the imaging surface (Figure 2B). This is not an option for prism-based TIRF setups, where the microscopy slide is the imaging surface due to background fluorescence.
NOTE: Alternatively, recycle the used microfluidic chambers16. For this, immerse the used chambers overnight in acetone (wrap the Coplin staining jar in aluminum foil to minimize evaporation) in the fume hood, and afterward sonicate for 5 min. Disassemble the chamber, and discard the coverslips and the stickers. If the disassembly does not work immediately, sonicate the chamber in acetone for 10 min more. Proceed with step 2.1.2. - Place the drilled quartz slides and approximately twice as many coverslips (since they may break easily) in a glass Coplin staining jar with a cover. Rinse 3x with ddH2O. Sonicate in ddH2O for 5 min, then rinse 3x with ddH2O.
- Sonicate in 10% laboratory glassware cleaning solution (see Table of Materials) for 30 min at 50 °C. Rinse at least 3x with ddH2O until the detergent bubbles are gone. Sonicate in ddH2O for 5 min. Rinse 3x with ddH2O.
- Sonicate in 1 M KOH solution for 30 min, then leave overnight. Rinse 3x with ddH2O. Sonicate in ddH2O for 5 min. Rinse 3x with ddH2O.
NOTE: Although corrosion due to excessive incubation is a concern previously raised by Chandradoss et al.15, we recommend this long incubation that enables avoiding Piranha etching. - Dry the slides and coverslips with N2(g).
- Treat the dried slides and coverslips with oxygen plasma cleaner for 30 min according to the manufacturer's instructions.
- Drill four holes in the quartz slides with a diamond driller according to the scheme given in Figure 2A to form two channels.
- Aminosilanization
- Prepare the 3% APTES-EtOH solution by thoroughly mixing 288.5 mL of absolute EtOH, 1.5 mL of ddH2O, and 9 mL of (3-aminopropyl)triethoxysilane (APTES) in a 500 mL Erlenmeyer flask.
- Place the clean slides and coverslips in a Coplin staining jar, immerse in the 3% APTES-EtOH solution, sonicate for 1 min, and incubate for 30 min.
- Rinse 3x with absolute EtOH, then 3x with ddH2O.
- Dry the slides and coverslips under N2(g) flow.
- Surface passivation and biotinylation
- Prepare a humid box by filling up an empty pipette tip box up to half with ddH2O. Place the slides inside the box, with the side to be treated facing up.
- Freshly prepare the bPEG-mPEG mixture in a 1.5 mL sterile-clean microcentrifuge tube by gently mixing 2 mg of biotin-polyethylene glycol-succinimidyl valerate 5000 (biotin-PEG-SVA) and 80 mg of methoxy polyethylene glycol-succinimidyl valerate (mPEG-SVA) in 640 µL of 100 mM sodium bicarbonate (NaHCO3) buffer, pH 8.3. Centrifuge the bPEG-mPEG mixture at 16,000 × g for 1 min to remove any air bubbles.
- Carefully remove the supernatant and add a 30 µL droplet to the center of the slide to cover both channels. Place an additional droplet on one side of the slide to have an extra PEGylated coverslip, as they break easily, and having a spare is useful. Finally, cover the drop with a clean coverslip, close the humid box, and perform the PEGylation overnight under dark conditions.
- Thoroughly rinse the PEG-passivated and biotinylated slides and coverslips with ddH2O. Notice the change in the hydrophobicity of the treated surfaces. Dry under N2(g) flow.
- Microfluidic chamber assembly
- Cut a double-sided sticker to create the channels. Attach the sticker to the slide, ensuring it covers the area of interest. Carefully place the coverslip on top, aligning it so that the PEGylated surfaces face each other.
- Place each assembled chamber in a 50 mL centrifuge tube and fill the tubes with N2(g), and store at -20 °C for up to 1 month.
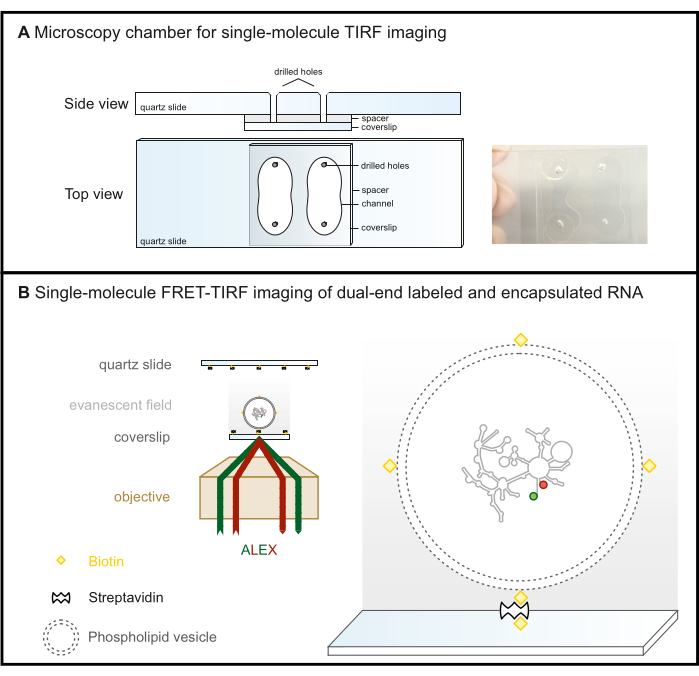
Figure 2: Single-molecule FRET-TIRF microscopy. (A) Microfluidic chamber for TIRF imaging. (B) FRET-labeled RNA is encapsulated in a biotinylated phospholipid vesicle and immobilized on a steptavidin-coated glass surface. This keeps the molecule of interest within the evanescent field (gray gradient), which is created by the incident light that is totally reflected at the critical angle in TIRF microscopy. Here, both fluorophores are subsequently excited with ALEX scheme. Abbreviations: FRET = Förster Resonance Energy Transfer; TIRF = Total Internal Reflection Fluorescence; ALEX = alternative laser excitation. Please click here to view a larger version of this figure.
3. Phospholipid vesicle encapsulation
- Lipid cake preparation
NOTE: Always handle chloroform under a fume hood.- Using a sterile needle, poke holes in the lid (from the inside out) of sterile-clean 2.0 mL microcentrifuge tubes, as shown in Figure 3A.
- Prepare the 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-cap biotinyl (bPE) stock solution by dissolving at least 2 mg of bPE in the appropriate amount of chloroform to a final concentration of 1 mg/mL.
- Prepare the 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) stock solution by dissolving at least 10 mg of DMPC in the appropriate amount of chloroform to a final concentration of 10 mg/mL.
- Prepare the bPE-DMPC mixture (keeping the 1:99 w/w ratio) by mixing 100 µL of bPE and 990 µL of DMPC stock solutions. Distribute 109 µL of the bPE-DMPC mixture per 2 mL tube.
- Place a cardboard cell partition insert of a microtube storage box in a Schlenk flask, serving as a tube holder. Using long tweezers, place the tubes containing the lipid mixture into the tube holder in a 500 mL Schlenk flask (Figure 3B).
NOTE: Make sure that they are upright and not tilted. - Evaporate the chloroform under a low flow of N2(g) overnight (or for at least 2 h until the solvent is completely evaporated).
- Seal the tubes with Parafilm to cover the holes. Store the lipid cakes at -20 °C for up to 1 month.
- RNA encapsulation
- Prepare the following buffers and solutions by adjusting the K+ and Mg2+ concentrations according to the biological system of interest.
- Prepare the 5x standard buffer (5x SB): 2.5 M KCl, 400 mM MOPS; adjust the pH to 6.9 with KOH, sterile filter, and store at 4 °C in the dark (respectively 1x SB: 500 mM KCl, 80 mM MOPS, pH 6.9).
- Prepare the antiblinking buffer (AB): 100 mM MgCl2, Trolox, 1x SB, sterile filter, and store at 4 °C in the dark for up to 1 week.
NOTE: Use a spatula tip of Trolox for 10 mL final volume, vortex to mix, and readjust the pH.
- Assemble the extruder using 100 nm polycarbonate (PC) membrane and 10 mm polyester (PE) drain disc, equilibrate the syringe and the membranes with AB, and heat to 30 °C (or essentially above the DMPC glass transition temperature of 24 °C).
- Mix 5 µL of 1 µM dual-end labeled RNA and 45 µL of AB (to a total volume of 50 µL).
- Hydrate the lipid cake with this RNA solution. At 30 °C, mix for 5 min by shaking at 1,400 rpm, then for 15 min at 700 rpm. Centrifuge for 2 min at 13,000 g and carefully transfer the supernatant into a new tube.
- Dilute the sample with 250 µL of AB.
- Fill the syringe with the RNA-lipid suspension. Extrude 35x at 30 °C on a heating block to encapsulate the dual-end-labeled RNA in phospholipid vesicles of 100 nm diameter.
NOTE: The size distribution of vesicles after using the extruder can be validated by dynamic light scattering (DLS), as shown in reference11.
- Prepare the following buffers and solutions by adjusting the K+ and Mg2+ concentrations according to the biological system of interest.

Figure 3: Lipid cake preparation. (A) The tube caps are poked to enable solvent evaporation. (B) Tubes containing the lipid mixture are placed in a Schlenk flask, and the chloroform is evaporated to obtain a lipid cake. Please click here to view a larger version of this figure.
4. smFRET-TIRF microscopy
- Surface immobilization
- Prepare the following buffers and solutions.
- Prepare the sugar-antiblinking buffer (SAB): 1% D-glucose (w/v) in AB, sterile filter, and store at 4 °C in the dark for up to 1 week.
- Prepare the 100x OSS: 100x Oxygen scavenging system (OSS): 3 mg of glucose oxidase (corresponding to 1.7 U), 10 µL of catalase (corresponding to 22 U) in 90 µL of 1x SB, store at 4 °C in the dark for up to 1 week.
- Freshly prepare the imaging buffer (IB) by mixing 198 µL of SAB and 2 µL of 100x OSS solution.
- Allow the microfluidic chamber to reach room temperature. Flush the chamber 2x with 200 µL of 1x SB.
- Fill the chamber with 50 µL of 20 µL/mL streptavidin solution and incubate for 5 min.
- Flush the chamber with 200 µL of 1x SB and then with 100 µL of AB.
- Immobilize the encapsulated RNA on the surface by adding 75 µL of vesicle suspension and incubate for 10 min.
- Flush the chamber with 200 µL of freshly prepared IB and incubate for 5 min.
- The chamber is now ready for data acquisition (Figure 2B). To prevent evaporation during long measurements, seal the holes as shown in Figure 2A. If leaks from channel to channel are of concern, seal the unused channel before starting the surface immobilization protocol.
NOTE: We recommend performing blank measurements as a quality check for the cleanliness of the microfluidic chambers and phospholipid vesicles against fluorescence contaminations.
- Prepare the following buffers and solutions.
We present the site-specific single- and dual-fluorescent labeling of the 915-nt RNA of interest, the yeast mitochondrial Sc.ai5γ group II intron, flanked by exonic sequences. The FRET fluorophore pair is positioned at the RNA ends via EDC/NHS activation of the 5'-phosphate and periodate oxidation of the 3'-ribose, followed by respective dye attachments. We then verified the RNA-dye conjugates by fluorescent gel electrophoresis, as presented in Figure 4A. The co-migration of RNA and the fluorophores on the agarose gel confirms the successful labeling. Next, as shown in Figure 4B, ensemble fluorescence spectroscopy was used to characterize the dual-labeled group II intron. Energy transfer, that is, FRET, was observed upon excitation of the donor dye proving the dual labeling of the RNA. Notably, consistent with the distance-dependent nature of FRET, the folding of the group II intron RNA in the presence of metal ions led to an increase in FRET efficiency, as evidenced by the decrease in donor emission (green arrow) and a corresponding increase in acceptor (red arrow) emission. This indicates that this FRET labeling tracks the conformational changes of the ribozyme.
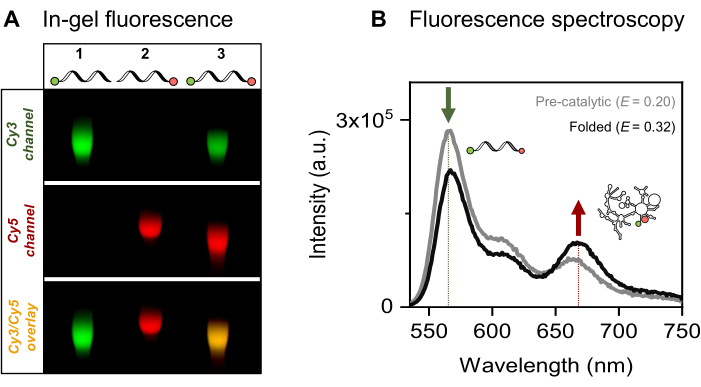
Figure 4: Characterization of the RNA-dye conjugates. (A) Analytical gel-based analysis of single- and dual-fluorescently labeled RNA shows the co-migration of the dyes with the RNA on a 2% agarose gel. Co-localization of the fluorophores in the dual-labeled sample is indicated by the yellow band in the merged image (bottom) of the Cy3 (top, green) and Cy5 (middle, red) channels, visualized under 532 nm and 635 nm illumination, respectively. Lane 1: 5'-sCy3 only labeled RNA, lane 2: 3'-Cy5 only labeled RNA, and lane 3: dual-end (5'-sCy3 and 3'-Cy5) labeled RNA. (B) Ensemble fluorescence spectroscopy confirms dual-labeling. Energy transfer upon donor excitation (λex = 515 nm, λem = 670 nm) verifies that both dyes are successfully coupled to the RNA. The gray curve represents the emission profile of the pre-catalytic RNA, while the black curve demonstrates increased FRET efficiency in the folded group II intron RNA (incubated with 500 mM KCl at 70 °C for 3 min, cooled to 42 °C for 5 min, followed by the addition of 100 mM MgCl2). This figure was adapted from Ahunbay et al.3 Please click here to view a larger version of this figure.
With the fluorescently dual-labeled wild-type group II intron in hand, we are now positioned to explore its dynamics at the single-molecule level. Once encapsulated in phospholipid vesicles, the labeled RNA is immobilized on a microscopy surface at very low surface densities to achieve single-molecule resolution for smFRET-TIRF. As seen in Figure 5A, several individual molecules can be tracked simultaneously. TIRF microscopy enables the real-time monitoring of FRET efficiency and its changes over time. Figure 5B exemplifies the static and dynamic single-molecule FRET traces of the labeled and encapsulated RNA. A typical dynamic trace exhibits anticorrelation between donor and acceptor signals that fluctuate the FRET efficiency. When the acceptor emission upon donor excitation increases, the donor emission correspondingly decreases, indicating a dynamic change in the inter-dye distance. This anticorrelation suggests conformational changes in the RNA molecule.
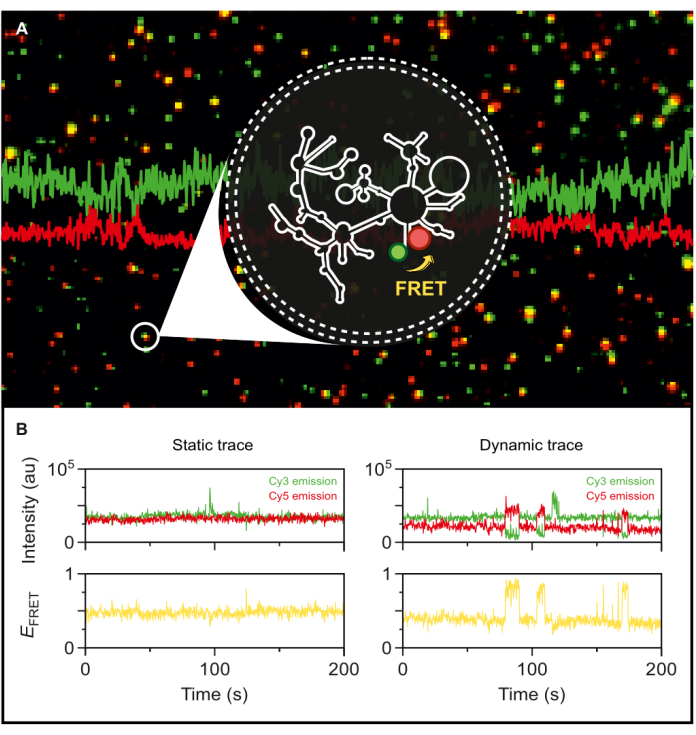
Figure 5: Highly dynamic behavior of group II intron RNA revealed by smFRET. The dual-end labeled group II intron RNA is encapsulated in a phospholipid vesicle and immobilized on the surface for imaging with an objective-based TIRF microscope. (A) Merged image of individual labeled RNA molecules exhibiting donor emission (sCy3, green) and acceptor emission (Cy5, red) upon 532 nm excitation. (B) The typical smFRET trajectories of (left) a static RNA molecule where the donor and acceptor intensities do not fluctuate over time and (right) a dynamic RNA molecule, where the donor and acceptor intensities anticorrelate, with FRET efficiencies in yellow. Single-molecules are localized and analyzed using MASH-FRET17. Direct excitation, bleed-through, and γ-factor corrections are applied. Abbreviations: smFRET = single-molecule Förster Resonance Energy Transfer. Please click here to view a larger version of this figure.
