In the study, we cloned the sgRNA target to Rorc and sgRNA-non-targeting coding sequences into pMX-U6-MCS vector with mCherry fluorescent protein (Figure 1A,B). Retrovirus production was carried out according to the protocol outlined in Figure 2. The transfection was initiated on day 0, and the retroviral harvest occurred on day 2. Transfection efficiency can be tested by the mCherry fluorescence intensity (Figure 1C). Before harvesting the virus, naïve CD4+ T cells were sorted and activated. These activated CD4+ T cells were then infected using spin infection, followed by continuation of the differentiation steps as described. Finally, the efficiency of transduction and the results of differentiation were evaluated using flow cytometry analysis and qPCR analysis.
Figure 3 illustrates the gating strategy applied for sorting naïve CD4+ T cells. After selecting the main cell population and singlet cells (as shown in the first two gates of Figure 3), FVD– CD4+ cells were gated to distinguish CD4 T cells in polarized to Th17 cells from those in APCs. Finally, IL-17A produced by CD4 T cells represent Th17 differentiation efficiency, which indicated the knockout efficiency. The percentage of mCherry-positive cells reflects the efficiency of retroviral infection, while the knockout efficiency is indicated by the percentage of IL-17A-positive cells. To enhance cell viability and minimize sorting duration, splenocytes were enriched using a mouse CD4+ T cell isolation kit, resulting in an enrichment efficiency surpassing 90%.
The efficiency of retrovirus infection, indicated by the presence of mCherry-positive cells, is typically around 40% (Figure 4A). Although infection efficiency can be increased through virus concentration, this approach comes at the expense of prolonged experimental duration and higher costs. In our experiments, infected cells cultured without the Th17 cell differentiation cocktail served as the Th17 differentiation negative control, designated as Th0. A non-targeting sgRNA was used as a positive control, referred to as sgNT. For gene editing, we employed gRNA targeting the RORγt gene (sgRNA-Rorc), which significantly reduced Th17 differentiation efficiency (Figure 4A). This intervention also led to a substantial decrease in the expression levels of Rorc and IL-17a (Figure 4B).
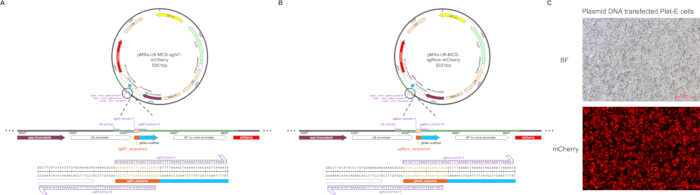
Figure 1: Example of sgRNA sub-cloned into a retroviral vector and transfection efficiency. The pMX-U6-MCS vector was used as the initial construct. The gRNA scaffold, along with the sgRNA sequences (the orange part) targeting the (A) scrambled site and (B) Rorc, were inserted downstream of the U6 promoter (the green part as a section for sequencing with U6 primers). The locations of the sgRNA-containing primers and sequencing primer sites are indicated below the circular vector The full names of the abbreviations on the plasmid are as follows. Abbreviations: AmpR = resistance to ampicillin, carbenicillin, and related antibiotics; LTR = long terminal repeat; MMLVψ = packaging signal of Moloney murine leukemia virus; gag = truncated Moloney murine leukemia virus (MMLV) gag gene lacking the start codon. (C) Microscopy imaging of mCherry in plasmid DNA-transfected Plat-E cells. Scale bar: 500 px. Please click here to view a larger version of this figure.
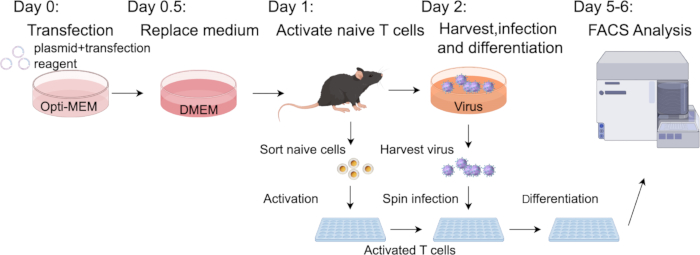
Figure 2: Schematic illustration of the protocol. The day before transfection, the density of the Plat-E cells needs to be adjusted to a proper range. The next day (day 0), perform transfection and replace medium 12 h later. On day 1, sort and activate the naïve T cells from Cas9-expressing mice. Subsequently (day 2), harvest the virus-containing culture supernatant and combine with the activated T cells for retroviral transduction. The infected cells are then differentiated into Th17 cells for 3–4 days and analyzed using FACS. Please click here to view a larger version of this figure.
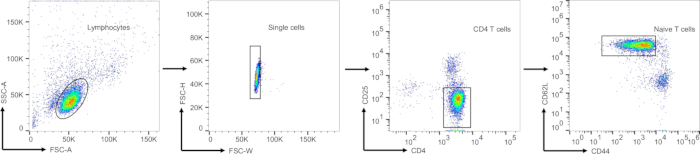
Figure 3: Gating strategy for sorting the CD4 naïve T cells. Surface staining was performed on the enriched CD4 T cells to label CD4, CD25, CD44, and CD62L. The gating strategy employed was as follows: initially, lymphocytes were gated based on forward scatter and side scatter parameters. Subsequently, single cells were identified using FSC-H and FSC-W. CD4+CD25– cells were then gated to exclude regulatory T cells (Tregs). Finally, CD44lowCD62Lhigh naïve T cells were identified. The isolated CD4+ naïve T cells were collected for subsequent experiments. Abbreviations: FSC = forward scatter; SSC = side scatter. Please click here to view a larger version of this figure.
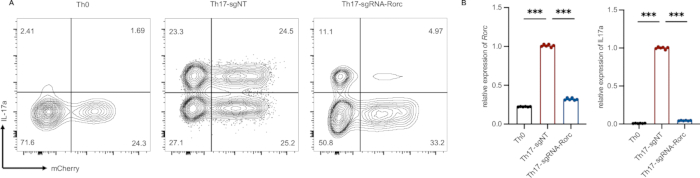
Figure 4: Example of successful transduction of activated T cells and Th17 differentiation. (A) The differentiation efficiency of Th17 is reduced significantly which is edited by RORγt gRNA (gating on mCherry+ IL-17a+population). (B) Rorγt and IL-17a relative mRNA expression in Th0 (infected cells cultured without Th17 cell differentiation cocktail), sgNT (non-targeting sgRNA) and sgRNA-Rorc (sgRNA targeting the RORγt gene). ***p < 0.001 (unpaired two-tailed student’s t-test; data are presented as mean ± SEM).Please click here to view a larger version of this figure.
| PCR Reaction Components | PCR Program | ||||
| Component | Volume (μL) | Step | Temp. | Time | Cycle |
| 2 × Phanta Mix (Dye Plus) | 25 | 1 | 95 ºC | 3 min | 1 |
| ddH20 | 20 | 2 | 95 ºC | 15 s | |
| Primer-F (10 μM) | 2 | 3 | 56 ºC | 15 s | |
| Primer-R (10 μM) | 2 | 4 | 72 ºC | 5 min 30 s | Go to Step 2 |
| 35 × | |||||
| Template (~30 ng) | 1 | 5 | 72 ºC | 5 min | 1 |
| 6 | 12 ºC | ∞ | |||
Table 1: PCR Reaction Components and PCR Program for gRNA vector construction.
