NOTE: All experiments are performed under BSL-2 condition. All materials that get in contact with Hepatitis E virus RNA or infectious virus must be rinsed properly with 4% Kohrsolin FF from a waste container inside the hood prior to disposal.
1. Plasmid preparation
- Inoculate 200 mL LB medium containing 100 µg/mL ampicillin with transformed Escherichia coli JM109 incorporating a plasmid encoding for the full-length HEV gt3 Kernow-C1p6 sequence (pBluescript_SK_HEVp6 [JQ679013]54 or pBluescript_SK_HEVp6-G1634R37). Incubate for 16 h at 37 °C under permanent agitation (170 rpm).
NOTE: Plasmid isolation was carried out using a plasmid extraction kit (see Table of Materials). - Following the manufactures protocol, spin down 200 mL of overnight culture at 6,000 x g and 4 °C for 10 min and discard the supernatant. The bacterial pellet can be frozen and stored at -20 °C.
- Resuspend the bacterial pellet in 4 mL of Resuspension buffer by pipetting up and down with a 10 mL serological pipette and a pipette man. Additionally, vortex rigorously.
- Add 4 mL of prewarmed Lysis buffer (30-40 °C). Invert gently for several times and incubate at room temperature (RT) for 5 min.
- Add 4.8 mL of Neutralization buffer, invert gently and make sure that the lysate is quantitatively neutralized (see manufacturer’s description). Then centrifuge at 11,000 x g and 4 °C for 20 min.
- Place a 2 cm x 2 cm mull piece inside a 1 mL non-filter tip, load resuspended, lysed and neutralized supernatant in 10 mL serological pipette, add 1 mL tip with mull to tip of the serological pipette and filter the supernatant in a new 50 mL tube.
NOTE: Supernatant can be stored at 4 °C for up to 30 min if necessary. - In several steps apply 750 µL of the filtered supernatant on a total of 4 filter columns (provided in the kit) and centrifuge at 11,000 x g for 30 s. Discard the flow-through and repeat this sequence until all supernatant is applied onto the filter columns.
- Wash each filter column twice with 500 µL of prewarmed (50 °C) wash buffer AW at 11,000 x g for 30 s and discard the flow-through afterwards.
- Wash each filter column with 600 µL of wash buffer A4 by centrifuging at 11,000 x g for 30 s. Discard the flow-through and dry the filter columns by centrifugation at 11,000 x g for 2 min.
- Elute the plasmid DNA from each filter column by transferring 60 µL of elution buffer onto the center of the filter columns. Incubate for 1 min at RT and centrifuge at 11,000 x g for 1 min.
- Combine eluates and measure the concentration of the extracted plasmid DNA using a spectrophotometer.
2. Linearization and DNA purification

Figure 1: Schematic experimental setup for the plasmid linearization and DNA purification. Please click here to view a larger version of this figure.
NOTE: The linearization increases RNA yield during in vitro transcription (step 3)
- To linearize the plasmid DNA (Figure 1) mix 10 µg of the template DNA (extracted in step 1), 10 µL of the buffer, 2 µL of MluI and adjust to a volume of 100 µL with H2O.
- Incubate for 1 h at 37 °C and confirm linearization of the plasmid by agarose gel electrophoresis (e.g., load non-digested and digested plasmid DNA [1 µL of DNA each] on a 1% agarose gel and run electrophoresis at 120 V constant current).
- Following the manufacturers’ protocol for DNA extraction (see Table of Materials, Figure 1), mix 500 µL of Binding buffer, provided in the kit, with 100 µL of linearized DNA. Apply the sample to a filter column, and centrifuge at 17,800 x g for 30 s. Discard the flow-through and place the filter columns back in the same tube.
- Wash the filter column by adding 650 µL of Wash buffer and centrifugation at 17,800 x g for 30 s. Discard the flow-through and place the filter column back in the same tube. To remove the residual Wash buffer, dry centrifuge at 17,800 x g for 60 s and place each filter column in a clean 1.5 mL microcentrifuge tube afterwards.
- Elute DNA by transferring 60 µL of prewarmed H2O (PCR grade, 70 °C) into the center of the filter. Incubate for 1 min at 70 °C and centrifuge at 18,000 x g for 1 min. Measure the concentration of the purified DNA using a spectrophotometer.
NOTE: Store DNA at -20 °C until in vitro transcription.
3. In-vitro transcription of full-length HEV genotype 3 p6 DNA and RNA purification
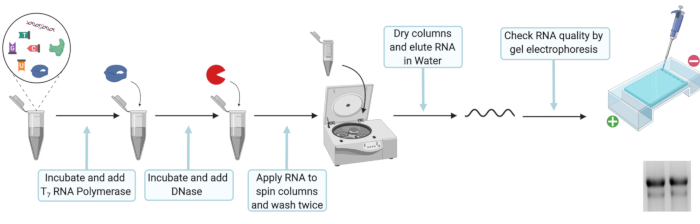
Figure 2: Schematic experimental setup for the in vitro transcription and RNA purification. Please click here to view a larger version of this figure.
NOTE: In vitro transcription is necessary to produce viral genomic RNA from plasmid DNA.
- For in vitro transcription mix 20 µL of 5x T7 transcription buffer, 10 mM DTT, 100 U of ribonuclease inhibitor, 25 mM ATP, CTP, and UTP, 12.5 mM GTP, 5 mM Ribo m7G Cap Analog, 2 µg of linearized DNA template, and 80 U of T7 RNA polymerase. Fill up to 100 µL with nuclease free H2O, mix well and incubate for 2 h at 37 °C.
NOTE: Short term storage at -20 °C is possible after step 3.1 to 3.1.2.- Add 2 µL of T7 RNA polymerase, mix well and incubate for another 2 h at 37 °C.
- To digest the initial DNA template, add 7.5 µL of DNase (RNase free, 1 U/µL), mix well and incubate at 30 min at 37 °C.
NOTE: Clean all surfaces and pipettes with surface decontaminant for RNase when working with RNA to avoid degradation. Also, ensure that all reaction tubes are RNase-free and sterile. Only use tips with filters and dilute solely with RNase-free and sterile water.
- Following the manufacture’s protocol for RNA extraction (see Table of Materials, Figure 2), prepare a premix of Lysis buffer and 100% ethanol by mixing 330 µL of Lysis buffer and 330 µL of 100% ethanol for each in-vitro transcription reaction. Add 660 µL of Lysis buffer-ethanol premix to 110 µL of RNA and vortex. Load sample on a filter column and centrifuge at 8000 x g for 30 s. Discard the flow-through and place the filter column back in the same tube.
- Add 600 µL of Wash buffer and centrifuge at 8000 x g for 30 s. Discard the flow-through and place filter column back in the same tube. Add 350 µL of Wash buffer and centrifuge at 8000 x g for 2 min to remove the residual wash buffer. Place each filter column in a clean 1.5 mL microcentrifuge tube. Open the lid of the filter column and let the column dry for 3 min.
- Elute RNA by placing 50 µL of RNase free H2O into the center of the filter column. Incubate for 1 min at RT and subsequentially centrifuge at 8,000 x g for 1 min. Check RNA integrity by loading 1 µL of RNA on to an agarose gel and measure the concentration of extracted RNA using a spectrophotometer.
NOTE: Store RNA at -80 °C until electroporation and exclusively thaw RNA on ice to avoid degradation.
4. Preparation of HepG2 cells for cell culture derived HEV (HEVcc) production
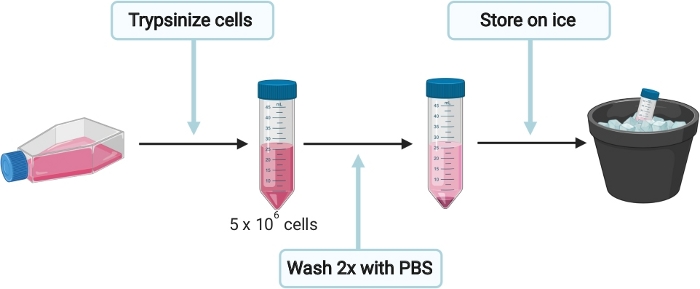
Figure 3: Schematic experimental setup for the preparation of HepG2 cells for cell culture derived HEV (HEVcc) production. Please click here to view a larger version of this figure.
NOTE: To avoid contamination, preparation of cells, electroporation, infection, harvesting and cell fixation were carried out under sterile conditions in a biosafety level 2 facility. Incubation steps at 37 °C that involve cells were accomplished in a 5% CO2 incubator.
- To prepare HepG2 cells for HEVcc production (Figure 3), seed cells in complete Dulbecco's Modified Eagle Medium (DMEM) (see Table 1) onto a 15 cm collagen (see Table 1, sterile filter PBS- acetic acid mix through 0.2 µm mesh before adding collagen) coated culture dish. Incubate at 37 °C until cells are 90% confluent.
NOTE: Each 15 cm dish containing 90% confluent cells will generate enough cells for up to 4 electroporation. - Gently remove the medium from plate and wash cells once with 10 mL of 1x PBS. Trypsinize cells by adding 3 mL of 0.05% trypsin-EDTA onto the cells and incubate at 37 °C until cells are detached completely. Resuspend cells in 10 mL DMEM complete medium and transfer the cell suspension into a 50 mL tube. Determine total number of cells.
- Since each electroporation requires 5 x 106 cells, transfer the appropriate volume in a new 50 mL tube and fill up to at least 35 mL with 1x PBS. Centrifuge cells at 200 x g for 5 min and carefully discard the supernatant without disturbing the cell pellet.
- Wash cells once again with 35 mL of 1x PBS at 200 x g for 5 min. Place cells on ice and do not remove 1x PBS yet, to keep the cells in suspension.
5. Electroporation of HepG2 cells
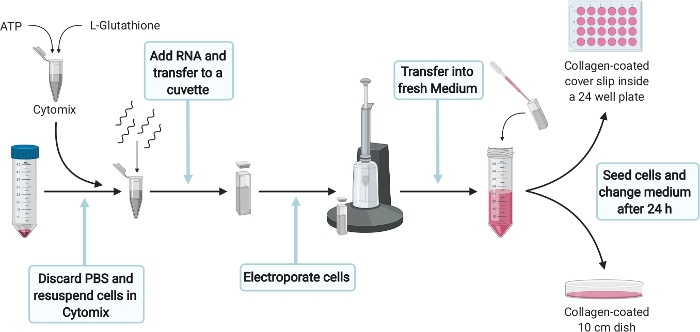
Figure 4: Schematic experimental setup for the electroporation of HepG2 cells. Please click here to view a larger version of this figure.
- For electroporation of HepG2 cells (Figure 4) prepare 400 µL of Cytomix complete per electroporation by supplementing 384 µL of Cytomix (see Table 1) with 2 mM ATP and 5 mM Glutathione. Prepare fresh right before use and place directly on ice.
NOTE: A correct yet quick execution of the following steps is crucial. Therefore, make sure that everything is prepared properly. - Carefully remove 1x PBS without disturbing the cell pellet from step 4.4. Resuspend 5 x 106 cells in 400 µL of Cytomix complete and add 5 µg RNA from step 3.2.2. Transfer the solution into a 4 mm cuvette and pulse once with 975 µF, 270 V for 20 ms with an electroporation system.
- After electroporation transfer cells as quickly as possible with a Pasteur pipette into 11 mL DMEM complete per electroporation.
NOTE: To make sure that RNA is successfully transfected into HepG2 cells, a transfection control (TC) will be performed. Expected transfection rates vary between 40-60% of ORF2-positive cells.
- After electroporation transfer cells as quickly as possible with a Pasteur pipette into 11 mL DMEM complete per electroporation.
- Transfer 10 mL of electroporated cells into a 10 cm culture dish coated with collagen. Add 1.3 x 105 electroprorated cells (300 µL) into one well of a collagen-coated 24-microtitre plate carrying a cover slip (later used as a transfection control). Distribute cells evenly and incubate at 37 °C.
- After 24 h, change the medium of the 10 cm dish and replace with 10 mL fresh DMEM complete. Do not change the medium of the transfection control. Incubate for another 6 days at 37 °C.
- Stop the transfection control in 24 well plate 5-7 d post electroporation depending on the cell density by continuing with the immunofluorescence staining protocol (step 8). The transfection efficiency is calculated by counting the number of ORF2 positive cells normalized to the total number of cells.
6. Harvesting of intra- and extracellular HEVcc
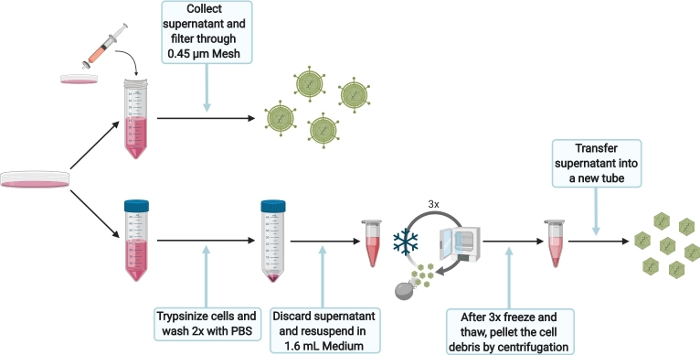
Figure 5: Schematic experimental setup for harvesting intra- and extracellular HEVcc. Please click here to view a larger version of this figure.
- To harvest extracellular HEVcc (Figure 5), filter the supernatant, obtained from the 10 cm dish after 6 days (step 5.3.1), through a 0.45 µm mesh to remove any cell debris. Store harvested extracellular HEVcc at 4 °C for the same day infection, otherwise store at -80 °C.
- To harvest intracellular HEVcc (Figure 5), wash cells with 1x PBS and trypsinize by adding 1.5 mL of 0.05% trypsin-EDTA. Incubate at 37 °C until cells are detached completely. Add 10 mL of DMEM complete, flush plate to detach cells and transfer cells suspension into 50 mL tube. Wash cells twice with PBS. Centrifuge at 200 x g for 5 min.
- Discard the supernatant and resuspend the cell pellet in 1.6 mL of DMEM complete per electroporation. Transfer the cell suspension into a 2 mL reaction tube.
NOTE: Do not use larger volumes as it would dramatically decrease viral loads. - Freeze (in liquid nitrogen) and thaw cells. Repeat this sequence 3 times.
NOTE: Do not pool more than one electroporation (1.6 mL) for the 3 freeze-and-thaw cycles as the lysis efficiency would be impaired. Do not vortex the cell suspension in between the cycles. Make sure to thaw cell suspension slowly (e.g., room temperature or on ice) to maximize viral loads. - High-speed centrifuge the lysed cells for 10 min at 10,000 x g to separate cell debris. Transfer the supernatant in a new tube. Take the supernatant for infection, otherwise store at -80 °C.
- Optionally, concentrate the extra- and intracellular HEVcc using a concentrator to increase viral loads (according to manufacturer’s protocol).
- Discard the supernatant and resuspend the cell pellet in 1.6 mL of DMEM complete per electroporation. Transfer the cell suspension into a 2 mL reaction tube.
7. Infection of HepG2/C3A cells with intra- and extracellular HEVcc
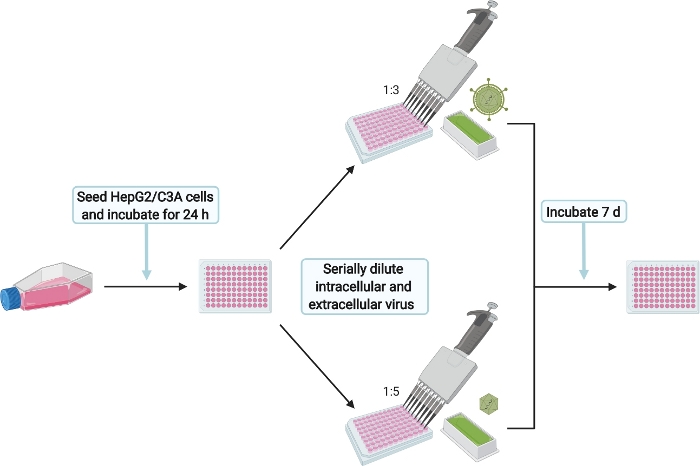
Figure 6: Schematic experimental setup for the infection of HepG2/C3A cells with intra- and extracellular HEVcc. Please click here to view a larger version of this figure.
NOTE: The infection of HepG2/C3A cells shall ensure that infectious particles were produced. Additionally, the titration of the harvested intracellular and extracellular HEVcc is used to calculate the virus titers in FFU/mL. This will be later referred to as infection control (IC).
- To prepare HepG2/C3A cells for HEVcc infection (Figure 6), prewarm Minimal Essential Medium (MEM) complete (see Table 1) to 37 °C.
- Seed 2 x 104 cells/well in 100 µL onto the collagen coated 96 well microtiter plate one day prior to step 6. Make sure to fill the outermost wells with 1x PBS to prevent evaporation of the medium inside the internal 60 wells. Incubate at 37 °C for 24 h.
- Infect with extracellular HEVcc by adding 50 µL of the supernatant (from step 6.1) to the HepG2/C3A cells seeded the day before. Mix well by pipetting up and down and serially dilute six times 1:3 by transferring 50 µL into the next well. Perform duplicates of serial dilution for technical replicates.
- Infect with intracellular HEVcc by adding 25 µL supernatant (from step 6.2.3) to the HepG2/C3A cells seeded the day before. Mix well by pipetting up and down and serially dilute six times 1:5 by transferring 25 µL into the next well. Perform duplicates serial dilution for technical replication.
- After 7 d at 37 °C stop the infection by continuing with the immunofluorescence staining protocol (step 8).
8. Immunofluorescence staining of transfection- and infection control
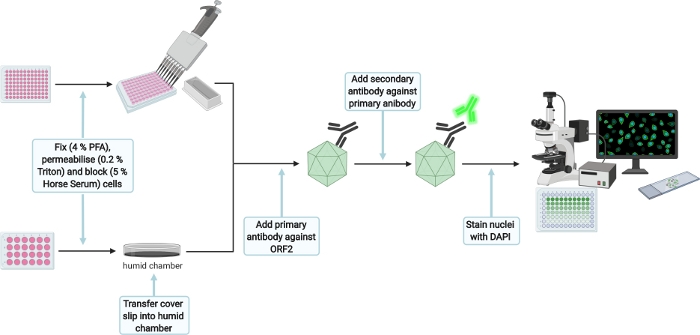
Figure 7: Schematic experimental setup for the immunofluorescence staining of transfection and infection control. Please click here to view a larger version of this figure.
- For immunofluorescence staining (Figure 7), wash 3x with 1x PBS and fix cells by dispensing 350 µL (transfection control [TC]) or 50 µL (infection control [IC]) of 4% Fixation solution per well. Incubate for 15 min at RT and carefully wash twice with 1x PBS afterwards.
NOTE: The protocol can be paused here for the transfection control until the infection of HepG2/C3A cells is stopped as well. Store well plate at 4 °C. Also seal with parafilm to prevent evaporation. - Permeabilize cells by adding 350 µL (TC) or 50 µL (IC) of 0.2% Triton X-100 (in 1× PBS) for 5 min at RT. Then carefully wash twice with 1x PBS and block with 350 µL (TC) or 50 µL (IC) of 5% Horse Serum (in 1x PBS) for 1 h at RT under constant agitation on an orbital shaker (30 rpm).
NOTE: Prepare small plastic dish (30 mm) by placing a moist tissue inside and a paraffin film layer on top, creating a humid chamber. Then transfer the TC cover slip facing up onto the paraffin film. The following steps for the TC will be executed in the humid chamber. - Add 70 µL (TC) or 25 µL (IC) of primary antibody anti-ORF2 8282 (HEV-specific rabbit hyperimmune serum,1:5,000 in 5% Horse Serum) per well and incubate overnight at 4 °C under constant agitation on an orbital shaker (30 rpm).
- Carefully wash twice with 1x PBS and add 70 µL (TC) or 25 µL (IC) of secondary antibody goat anti-rabbit 488 (1:1,000 in 5 % Horse serum). Incubate for 1 h under constant agitation on an orbital shaker (30 rpm) in the dark. Again, carefully wash twice with 1x PBS
- Add 70 µL (TC) or 25 µL (IC) of DAPI (1:10,000 in H2O) and incubate for 1 min. Carefully wash twice with H2O. Take out the cover slip from the humid chamber, mount cover slip with 6 µL of the mounting reagent upside down on a cover slide (only for TC) and let mounting reagent dry for 16 h at RT in the dark. Store IC in water until imaging and seal the plate with paraffin film to avoid drying due to evaporation.
- Take images to confirm successful transfection and infection.
NOTE: Comparable results were obtained using the commercially available anti-ORF2 1E6 antibody57 (1:200 in 5 % Horse serum) and a secondary antibody donkey anti-mouse (1:1,000 in 5 % Horse serum)
9. FFU determination
NOTE: One FFU is defined as one or more ORF2-positive cells separated from another FFU by at least three negative cells.
- For extracellular HEVcc start to count all ORF2-positive foci of the two wells in the two lowest dilutions. A total of four wells should be counted. The focus forming units (FFU) per milliliter can be calculated with the following equation:
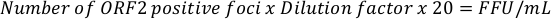
NOTE: Expected titers vary between 102 and 5 x 104 FFU/mL. - For intracellular HEVcc start to count all ORF2-positive foci in wells where between 50 to 100 foci are observed. Additionally, count the two wells in the next higher dilution. A total of four wells should be counted. The focus forming units (FFU) per milliliter can be calculated with the following equation:

NOTE: Expected titers vary between 105 and 3 x 106 FFU/mL. The factors 20x and 40x are used to extrapolate the number of FFU per 50 µL and 25 µL to 1 mL and can be adapted accordingly.
In this protocol, we describe the production of high titer infectious HEVcc. The first step is to isolate plasmid DNA (pBluescript_SK_HEVp654 and pBluescript_SK_HEVp6-G1634R37, Figure 8a), which then is linearized by restriction digestion and purified for in vitro transcription (Figure 1). A successful linearization can be verified by comparing the non-digested plasmid-DNA to the digested plasmid-DNA using gel electrophoresis. In addition to a size-shift, only one DNA band should be visible representing the linear form. The linearization is complete when the two other bands above and below the linear form, representing the nicked circle and the supercoiled form, respectively, are completely diminished (Figure 8b). The yield of the purified DNA should exceed 150 ng/µL. Only if these characteristics hold true the linearized DNA should be used for in vitro transcription. The in vitro transcribed RNA should be as well checked using gel electrophoresis and in case of low RNase abundancy should show distinct bands rather than a blurred smear (Figure 8c). Additionally, the purified RNA yield should exceed 500 ng/µL The in vitro transcribed RNA (Figure 2) eventually is electroporated into HepG2 cells for virus production (Figure 3 and Figure 4). Successful electroporation is monitored by the immunofluorescence staining of the transfection control (Figure 9a). The transfection efficiency should exceed 40% (Figure 9b). A replication deficient mutant serves as a negative control, to ensure specificity of the ORF2 staining, as no ORF2 expression is expected (Figure 9c). After 7 days of incubation the enveloped (extracellular) HEVcc are harvested by collecting and filtering the cell culture supernatant. Non-enveloped (intracellular) HEVcc is released from the cells by several freeze and thaw cycles. To remove any cell debris the cell lysate is centrifuged at high speed (Figure 5). Subsequently, both HEVcc species are utilized to infect HepG2/C3A cells by serial dilution (Figure 6 and Figure 9d). According to the equations above (see step 9) viral titers are determined by FFU calculation.
Representative results are depicted in Figure 9. The transfection control should comprise around 50% ORF2-positive cells to guarantee an efficient amount of virus being produced (Figure 9a,b). The less ORF2-positive cells the lower the titer will be. Precisely following the steps mentioned in the protocol will generate titers that vary between 105 and 3 x 106 FFU/mL for the non-enveloped (intracellular) HEVcc. For the enveloped (extracellular) HEVcc titers between 102 and 5 x104 FFU/mL are expected (Figure 9e). Additionally, elevated FFU counts were observed for the G1634R mutant. When calculating the ratio between genome copies and infectious viral particles the produced intracellular HEVcc for both p6_WT and p6_G1634R was found to be lower compared to the extracellular HEVcc, suggesting a higher specific infectivity of the non-enveloped HEV species (Figure 9f).

Figure 8: Generation of in vitro transcribed RNA.
(A) Example of an absorbance spectrum of isolated Plasmid-DNA. (B) Gel electrophoresis of non-digested and digested Plasmid-DNA. (C) Gel electrophoresis of in vitro transcribed RNA. Please click here to view a larger version of this figure.
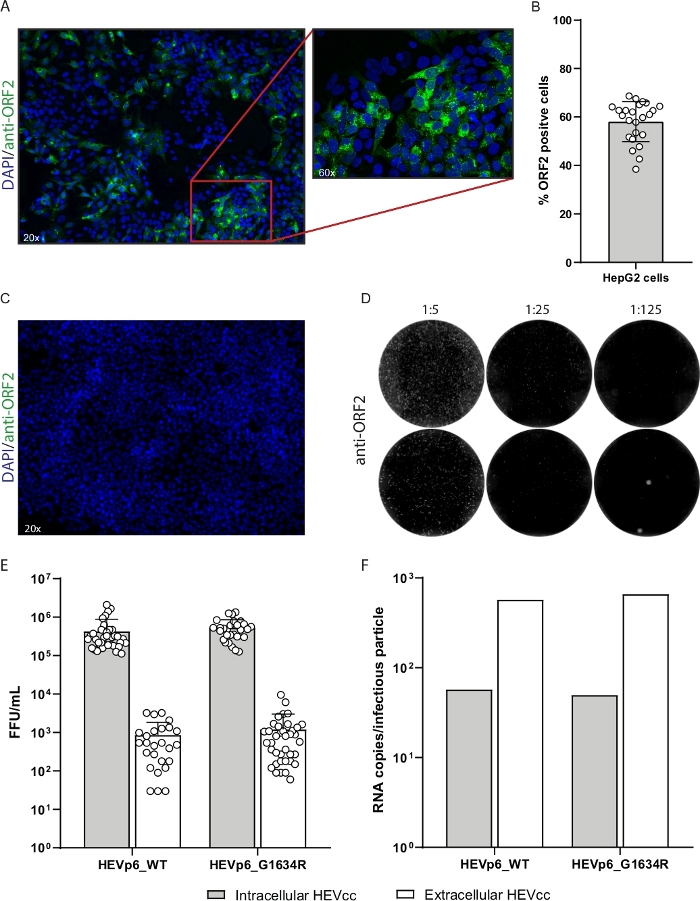
Figure 9: Representative results of high titer HEVcc production.
(A) Transfection control of electroporated HepG2 cells. Transfected cells were stained with anti-ORF2 antibody (green, LS-Bio) and DAPI (blue). (B) The transfection efficiency was calculated by the number of ORF2-positive cells normalized to the number of total cells. (C) A replication-deficient mutant serves as a negative control for immunofluorescence staining, to ensure ORF2 specificity. (D) For FFU determination serial dilutions of the produced virus stocks were performed. ORF2 positive cells are depicted in white. (E) Viral titers were calculated by FFU counting and (F) viral loads were determined by qPCR and normalized to FFU/mL. Bars show the mean and standard deviation of 38 and 10 independent experiments, respectively. Please click here to view a larger version of this figure.
| Working solution | Concentration | Volume |
| Collagen | 40 mL | PBS |
| 40 µl | Acetic acid | |
| 1 mL | Collagen R solution 0.4% sterile | |
| Cytomix | 120 mM | KCl |
| 0.15 mM | CaCl2 | |
| 10 mM | K2HPO4 (pH 7.4) | |
| 25 mM | HEPES | |
| 2 mM | EGTA | |
| DMEM complete | 500 mL | DMEM |
| 5 mL | Pen/Strep | |
| 5 mL | MEM NEAA (100X) | |
| 5 mL | L-Glutamin | |
| 50 mL | Fetal bovine serum | |
| MEM complete | 500 mL | MEM |
| 5 mL | Sodium Pyruvat | |
| 5 mL | Gentamycin | |
| 5 mL | MEM NEAA (100X) | |
| 5 mL | L-Glutamine | |
| 50 mL | ultra-low IgG |
Table 1: Table of Buffer Composition.
