1. Preparation of the Cas12a plasmids
NOTE: The plasmid containing the Lachnospiraceae bacterium ND2006 Cas12a (LbCpf1, pCSN067) codon optimized for expression in S. cerevisiae, was previously constructed19, deposited at a plasmid repository (see the Table of Materials). This is a single-copy episomal S. cerevisiae/E. coli shuttle plasmid containing a KanMX resistance marker gene to allow for selection of S. cerevisiae transformants on geneticin (G418).
- Obtain the pCSN067 plasmid (see the Table of Materials).
- Amplify the pCSN067 plasmid to obtain a high amount.
- Transform 25 µL of purchased chemically competent E. coli cells with the plasmid pCSN067 according to the manufacturer’s protocol. Dilute the transformation mix 10 and 50 times in 2x peptone-yeast (PY). Plate out 10x and 50x dilutions on 2x PY agar plates containing ampicillin (0.1 g/L) and incubate overnight at 37 °C.
- Pick 2 to 3 colonies and inoculate each colony in 3 mL of 2x PY and grow overnight at 37 °C in a shaking incubator at 180 rpm.
- Purify the plasmid using a plasmid purification kit according to manufacturer's instructions.
2. Preparation of the single crRNA array expression cassette
- Prepare the single crRNA array.
NOTE: The single crRNA array comprises an SNR52 RNA polymerase III promoter from S. cerevisiae2, a direct repeat specific for LbCas12a and a spacer (genomic target sequence), together repeated for each target19 and ends with a SUP4 terminator from S. cerevisiae2. The single crRNA array is assembled by in vivo recombination into the linearized plasmid pRN1120 to generate a circular plasmid, thus regions homologous to plasmid pRN1120 must be present at the start and end of the single crRNA array (see Figure 2A). It is recommended to in advance evaluate the functionality of a number of designed crRNAs separately19. This information is subsequently used to select most functional crRNAs to combine these into the direct repeat and spacer sequences to create a single crRNA array for the multiplexing purpose.- Order the single crRNA array for multiplex genome editing experiments as synthetic DNA (see the DNA sequence of the single crRNA array in Supplementary Table 1).
- Amplify the ordered single crRNA array (e.g., using primers KC-101 and KC-102 (Supplemental Table 2)). Prepare the PCR amplification mix containing: 0.5 µL of DNA polymerase, 10 µL of 5x buffer required for the DNA polymerase, 1 µL of 10 mM dNTPs, 2.5 µL of 10 µM forward primer, 2.5 µL of 10 µM reverse primer, 2 µL of DNA template at a concentration of 5 ng/µL and ultrapure H2O up to a total volume of 50 µL.
- Perform the reaction in a thermocycler using the following program: (i) 98 °C for 3 min, (ii) 98 °C for 10 s, (iii) 60 °C for 20 s, (iv) 72 °C for 15 s – repeat steps (ii) to (iv) 30 times, (v) 72 °C for 5 min (vi) hold at 12 °C until further analysis.
- Analyze the PCR products by electrophoresis by running the samples on a 0.8% agarose gel at 5 V/cm for 40 min using a DNA loading dye and DNA ladder with DNA fragments in a range of 100 to 10,000 bp.
- Purify the PCR products using a PCR purification kit according to the instructions of the manufacturer.
- Prepare the single crRNA array recipient plasmid.
NOTE: The single crRNA array is expressed from the S. cerevisiae/E. coli shuttle plasmid pRN112019 (see the Table of Materials). This multi-copy plasmid contains a NatMX resistance marker gene to allow selection of S. cerevisiae transformants on nourseothricin (NTC).- Obtain the pRN1120 plasmid.
- Amplify the pRN1120 plasmid to obtain a high amount.
- Transform 25 µL of purchased chemically competent E. coli cells with plasmid pRN1120 according to the manufacturer’s protocol. Dilute the transformation mix 10 and 50 times in 2x PY. Plate out 10x and 50x dilutions on 2x PY agar plates containing ampicillin (0.1 g/L) and incubate overnight at 37 °C.
- Pick 2 to 3 colonies and inoculate each colony in 3 mL of 2x PY and grow overnight at 37 °C in a shaking incubator at 180 rpm.
- Purify the plasmid using a plasmid purification kit according to the manufacturer's instructions.
- Linearize plasmid pRN1120 with EcoRI-HF and XhoI. For this, prepare a digestion mix composed of 1 µg of pRN1120, 5 µL of 10x buffer (1x buffer contains 50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 100 µg/mL bovine serum albumin [BSA]; pH 7.9), 1 µL of EcoRI-HF (20 U), 1 µL of XhoI (20 U) and ultrapure H2O up to a total volume of 50 µL. Incubate the digestion mix at 37 °C for 2 h and inactivate at 65 °C for 20 min.
- Analyze the linearized plasmid by electrophoresis on an agarose gel (0.8%, 40 min, 5 V/cm) using a DNA loading dye and DNA ladder with DNA fragments in a range of 100 to 10,000 bp. As a control include a circular plasmid in the analysis.
- Purify the linearized plasmid using a PCR purification kit according to the instructions of the manufacturer.
3. Preparation of Promoter-ORF-Terminator (POT) donor DNA constructs
- Order a set of promoter (P) of different strength, open reading frame (O) and terminator (T) sequences as synthetic DNA such that each element contains standardized 4-bp recognition sequences that are flanked by BsaI sites to enable Golden Gate Cloning (GGC) assembly26 (see the detailed designs in Supplementary Table 3 and sequences in Supplementary Table 4).
- Assemble POT expression cassettes composed of a promoter, open reading frame, terminator and connectors sequences via a 4-part assembly using a GGC reaction21, into a destination vector that already contains pre-specified 50-bp connectors sequences (see Supplementary Table 4 and references26,27).
- Measure the concentration of DNA parts using a spectrophotometer. Dilute each DNA part in ultrapure H2O to a final concentration of 15 fmol/µL.
- Prepare a reaction mix composed of DNA fragments: 2 µL of promoter, 2 µL of open reading frame, 2 µL of terminator and 2 µL backbone (Level 1 destination vectors as described in 26), 4 µL of 5x T4 DNA ligase buffer, 2.5 µL of 1 U/µL T4 DNA Ligase, 1.5 µL of 20 U/µL BsaI-HF and ultrapure H2O up to a total volume of 20 µL.
- Perform the GGC reaction in a thermocycler using the following program: (i) 37 °C for 2 min, (ii) 16 °C for 5 min – repeat steps (i) and (ii) 50 times, (iii) 50 °C for 60 min, (iv) 80 °C for 45 min, (v) hold at 12 °C until further analysis.
- Transform 25 µL of purchased chemically competent E. coli28 cells with 3 µL of the GGC reaction mix according to manufacturer’s protocol. Dilute the transformation mix 10 and 50 times in 2x PY. Plate out 10x and 50x dilutions on 2x PY agar plates containing ampicillin (0.1 g/L) and incubate overnight at 37 °C.
- Pick 2 to 3 colonies and inoculate each colony in 3 mL of 2x PY and grow overnight at 37 °C in a shaking incubator at 180 rpm.
- Purify the plasmids using a plasmid purification kit according to manufacturer's instructions.
- Check if POT expression cassettes were assembled correctly in the GGC reaction by PCR.
- Design primers complementary to the connector sequence present at the start and the end of each expression cassette (see Figure 2B). For connectors chosen in this protocol use primers KC-103 to KC-108 (see Supplementary Table 2).
- Prepare PCR amplification mixes for each plasmid containing: 0.5 µL of proofreading DNA polymerase, 10 µL of 5x buffer required for the DNA polymerase, 1 µL of 10 mM dNTPs, 2.5 µL of 10 µM forward primer, 2.5 µL of 10 µM reverse primer, 2 µL of DNA template with a concentration of 5 ng/µL, and ultrapure H2O up to a total volume of 50 µL.
- Perform the PCR reaction in a thermocycler using the following program: (i) 98 °C 3 min, (ii) 98 °C for 10 s, (iii) 60 °C for 20 s, (iv) 72 °C for 2 min 30 s – repeat steps (ii) to (iv) 30 times, (v) 72 °C for 5 min, (vi) hold at 12 °C until further analysis.
NOTE: Resulting PCR products consist of 50-bp of the 5’ connector, promoter, open reading frame, terminator and 50-bp of the 3’ connector.
- Analyze the PCR products by electrophoresis by running samples on a 0.8% agarose gel at 5 V/cm for 40 min using a DNA loading dye and DNA ladder with DNA fragments in a range of 100 to 10,000 bp.
4. Preparation of integration flank DNA sequences containing connectors sequences
- Purify genomic DNA from wild type S. cerevisiae CEN.PK113-7D29.
- Grow the strain in a 500 mL shake flask filled with 100 mL of yeast extract peptone dextrose (YEPD, 2% glucose) medium at 30 °C and shaking at 250 rpm for 48 hours.
- Harvest the cells by centrifugation of 2 mL of broth at 16,000 x g for 1 min and discard the supernatant.
- Resuspend the cells in physiological salt (200 µL; 0.85% NaCl solution) with RNase (10 µL, 10 mg/mL) and yeast lytic enzyme (4 µL). Incubate the cell suspension at 37 °C for 15 min.
- Add 300 µL of cell lysis solution (see Table of Materials) and vortex shortly.
- Add 168 µL of protein precipitation solution (see Table of Materials) and vortex vigorously for 20 s.
- Separate the protein fraction by centrifugation at 16,000 x g and 4 °C for 10 min. Collect 600 µL of supernatant in a new tube and mix with 600 µL of isopropanol and vortex shortly.
- Recover DNA by spinning down at 16,000 x g at room temperature for 10 min. Discard the supernatant and keep the pellet.
- Wash the pellet with 200 µL of ethanol (70%). Centrifuge at 16,000 x g at room temperature for 10 min and remove the supernatant. Evaporate the ethanol by incubating the tube at room temperature for 10 min with the lid opened.
NOTE: If liquid in the tube is still visible, repeat the step 4.1.8. Do not dry the pellet for longer than 10 min to prevent decreased solubility of the DNA. - Dissolve DNA in 50 µL of TE buffer. Store purified DNA at 4 °C.
- For each integration site, design integration flank DNA sequences (approx. 500 bp) such that approximately 1000 bp of genomic DNA will be removed upon introduction of donor DNA (see the schematic design in Figure 2B and sequences in Supplementary Table 4).
- Design primers to generate the flanking regions by PCR.
- For the left flanking region, design forward and reverse primers to amplify approximately 500 bp of the genomic DNA region positioned 5’ (left) of the integration site of interest.
NOTE: The forward primer includes 20 bp of homology with the intended flanking region. The reverse primer includes 20 bp with homology with the intended flanking region and contains the desired 50-bp connector sequence to enable in vivo assembly in the Cas12a editing on the genome later on. - For the right flanking region, design forward and reverse primers to amplify approximately 500 bp of the genomic DNA region positioned 3’ (right) of the integration site of interest.
NOTE: The forward primer includes 20 bp with homology with the intended flanking region and contains the desired 50-bp connector sequence to enable in vivo assembly in the Cas12a editing on the genome later on. The reverse primer includes 20 bp of homology with the intended flanking region.
- For the left flanking region, design forward and reverse primers to amplify approximately 500 bp of the genomic DNA region positioned 5’ (left) of the integration site of interest.
- Amplify the flanking regions with the designed primers (e.g., primers KC-109 to KC-120 enclosed in Supplementary Table 2).
- Measure the concentration of purified genomic DNA that will serve as the template in the PCR. Adjust the DNA concentration to 50 ng/µL.
- Prepare PCR amplification mixes composed of genomic DNA (1 – 4 µL of 50 ng/µL genomic DNA dilution) purified in step 4.1, forward and reverse primer (10 µM each), 1 µL of 10 mM dNTPs, 10 µL of 5x buffer required for the DNA polymerase, 0.5 µL of DNA polymerase (1.0 U), and ultrapure H2O up to total volume of 50 µL.
- Perform PCRs in a thermocycler using the following program: (i) 98 °C for 3 min, (ii) 98 °C for 20 s, (iii) 60 °C for 20 s, (iv) 72 °C for 15 s, repeat steps (ii) to (iv) 30 times, (v) 72 °C for 5 min, (vi) hold at 12 °C until further analysis.
- Analyze the PCR products by electrophoresis on a 0.8% agarose gel at 5 V/cm for 40 min using a DNA loading dye and DNA ladder with DNA fragments in a range of 100 to 10,000 bp.
- Purify the correct PCR products using a PCR purification kit according the instructions of the manufacturer.
5. Transformation to S. cerevisiae
NOTE: Perform transformation using a protocol based on the methods developed by Gietz et al. (1995)30 and Hill et al.31 which can be used for various strains of S. cerevisiae. The protocol described below is sufficient for 1 transformation.
- Prepare solutions required for transformation.
- Prepare the following stock solutions and filter-sterilize: 10x TE buffer containing 100 mM Tris-HCl (pH 7.5), 10 mM EDTA, total volume of 50 mL; 1 M LiAc at pH 7.5, total volume of 50 mL; 50% PEG 4000, total volume of 100 mL.
NOTE: Always check that PEG 4000 stock is at pH 5. This stock should not be stored longer than one month. - Prepare the following solutions using stocks: Prepare LiAc-TE solution containing 0.1 M LiAc, 10 mM Tris-HCl, 1 mM EDTA, total volume of 0.5 mL. Prepare PEG-LiAc-TE solution containing 40% PEG 4000, 0.1 M LiAc, 10 mM Tris-HCl, 1 mM EDTA, total volume of 1 mL.
NOTE: It is crucial for successful transformation that PEG-LiAc-TE and LiAc-TE solutions are freshly prepared.
- Prepare the following stock solutions and filter-sterilize: 10x TE buffer containing 100 mM Tris-HCl (pH 7.5), 10 mM EDTA, total volume of 50 mL; 1 M LiAc at pH 7.5, total volume of 50 mL; 50% PEG 4000, total volume of 100 mL.
- First transformation round (prepare the strain pre-expressing Cas12a).
NOTE: In all the transformation steps, use water with a pH higher than 5. It is recommended to use demineralized water in all the steps of the transformation.- Prepare a pre-culture by growing strain CEN.PK113-7D in a 100 mL shake flask containing 20 mL of YEPD (2% glucose) medium and incubate overnight at 30 °C with shaking at 250 rpm.
- Measure the OD600 of the pre-culture (ODpc). Calculate the dilution factor (df) between the volume of pre-culture and the volume of fresh medium required for preparation of the cells pre-expressing Cas12a to be used in the transformation (transformation culture). In the calculations assume the optical density of the transformation culture (ODtc) to be 1.0 after the incubation step described in 5.2.3 (ti).
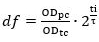
where ti and τ are the incubation time and doubling time, respectively.- Calculate the volume of the pre-culture (Vi) required for inoculation of the transformation culture (Vtc) based on the dilution factor.
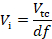
- Calculate the volume of the pre-culture (Vi) required for inoculation of the transformation culture (Vtc) based on the dilution factor.
- Prepare the transformation culture by inoculation of 20 mL of YEPD (2% glucose) (Vtc) with the volume of pre-culture determined in the previous step (Vi). Incubate at 30 °C with shaking at 250 rpm.
- Measure the OD600 of the transformation culture until an OD600 of 1.0 is reached.
- Harvest the cells by centrifugation of the 20 mL broth at 2,500 x g for 5 min. Discard the supernatant and wash the cells in 20 mL of room temperature demineralized water. Repeat the centrifugation step and keep the cell pellet.
- Resuspend the cells in 100 µL of LiAc-TE solution and transfer to a microcentrifuge tube.
- Add 5 µL of single-stranded carrier DNA (10 mg/mL salmon sperm DNA) and mix by pipetting.
- Pipette 1 µg of plasmid pCSN067 to the microcentrifuge tube.
NOTE: The total volume of the DNA mixture should not exceed 100 µL to prevent a lower transformation efficiency. - Add 600 µL of PEG-LiAc-TE solution and mix by pipetting. Incubate for 30 min at 30 °C while shaking at 450 rpm in a table top heat block.
- Add 70 µL of DMSO (100%) to the transformation mixture and mix by pipetting. Perform heat-shock by incubating the transformation mixture at 42 °C for 15 minutes in a water bath.
- Recover the cells by transferring the mixture to a 15 mL round bottom tube and add 10 mL of YEPD (2% glucose) to the tube. Incubate overnight at 30 °C with shaking at 250 rpm.
- Centrifuge the transformation mix at 2,500 x g for 5 min. Discard the supernatant and resuspend the cell pellet in approximately 200 µL of the remaining solution.
- Plate out 150 µL of the transformation mix and a 20x dilution in YEPD (2% glucose) of transformation mix on YEPD (2% glucose) agar plates supplemented with 0.2 g/L G418. Incubate the plates at 30 °C for 48 – 72 hours.
- Pick a single transformant and re-streak on a YEPD (2% glucose) agar plate supplemented with 0.2 g/L G418 to obtain single colonies.
- Second transformation round (perform multiplex genome editing with CRISPR/Cas12a).
- Prepare a pre-culture by growing the strain pre-expressing Cas12a, created in the first transformation round (step 5.2), in a 100 mL shake flask containing 20 mL of YEPD (2% glucose) medium supplemented with 0.2 g/L G418. Incubate overnight at 30 °C with shaking 250 rpm.
NOTE: For multiple transformations, adapt the volume of the pre-culture. - Follow the steps 5.2.2 to 5.2.7 for the first transformation round.
NOTE: For multiple transformations, adapt the volumes of required solutions and culture of the strain pre-expressing Cas12a. - Pipette 1 µg of the single crRNA array, 1 µg of the linearized recipient plasmid for the crRNA array, 1 µg of donor DNA and 1 µg of each flanking region (step 4.3) in the microcentrifuge tube.
NOTE: The total volume of the DNA mixture should not exceed 100 µL to prevent a lower transformation efficiency. - Prepare the following controls for the transformation: negative control (ultrapure H2O); positive control for determination of the transformation efficiency (1 µg of circular pRN1120); a control verifying if introduction of donor DNA is conducted via CRISPR editing (1 µg of circular pRN1120, 1 µg of all donor DNA expression cassettes and 1 µg of flanking regions but no single crRNA array); control verifying if donor DNA can be integrated outside of target (1 µg of linearized pRN1120, 1 µg of donor DNA expression cassettes and 1 µg of the single crRNA array but no flanking regions); a control verifying full linearization of pRN1120 (1 µg of linearized pRN1120).
- Follow the steps 5.2.9 to 5.2.12 for the first transformation round.
- Plate out 150 µL of the transformation mix and 20x dilution in YEPD (2% glucose) of transformation mix on YEPD (2% glucose) agar supplemented with 0.2 g/L G418 and 0.2 g/L NTC. Plate out controls on YEPD (2% glucose) agar supplemented with the appropriate selection (G418 and/or NTC or no selection). Incubate the plates at 30 °C for 48 – 72 hours.
- Pick a single colored transformant and re-streak on a YEPD (2% glucose) agar plate to obtain single colored colonies.
- Prepare a pre-culture by growing the strain pre-expressing Cas12a, created in the first transformation round (step 5.2), in a 100 mL shake flask containing 20 mL of YEPD (2% glucose) medium supplemented with 0.2 g/L G418. Incubate overnight at 30 °C with shaking 250 rpm.
6. Evaluation of the genome editing efficiency
- Count the number of colored colonies and white colonies on the transformation plates.
- Calculate genome editing efficiency by dividing the number of colored colonies by the total number of colonies (both white and colored), as shown in Table 1.
7. Confirmation of integration of donor DNA at the intended loci
- Re-streak a colored single colony from a transformation plate on a YEPD (2% glucose) agar plate without G418 and NTC selection and incubate for 48 hours at 30 °C.
- Pick a single colony and inoculate a 500 mL shake flask filled with 100 mL of YEPD (2% glucose) medium. Incubate for 48 hours at 30 °C and shaking at 250 rpm.
- Isolate the genomic DNA as described in Section 4.1.
NOTE: Alternatively, use a protocol for preparation of yeast for colony PCR previously proposed by Looke et al.32. In this case, growth in liquid medium (Section 7.2) can be skipped. - Verify correct integration by amplification of two fragments per integrated expression cassette.
- Design primers which anneal to genomic DNA outside of the transformed flanking regions and the gene of interest (see examples in Supplementary Table 2, KC-121 to KC-132). When using primers KC-121 to KC-132, set the annealing temperature in the PCR program to 62 °C.
- Amplify region of interest as described in Section 4.4.2. Adapt the PCR program, specifically adjust the time of the extension step in PCR according to the length of the template and manufacturer’s recommendations for the DNA polymerase.
- Check the size of the PCR products by electrophoresis on an agarose gel (0.8%, 40 min, 5 V/cm) using a DNA loading dye and DNA ladder with DNA fragments in a range of 100 to 10,000 bp.
8. Creation of yeast pixel art using an acoustic liquid handler
- Prepare a picture template for the yeast pixel art.
- Resize the original RGB picture (220 × 280 pixels, see the representative results), e.g. using ImageJ to create a final 64 × 96 pixels (width × height) grey-scale image visualized in intended colors (Representative Results).
- Convert the RGB picture into grey-scale using this formula:
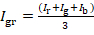
where Igr, Ir, Ig, Ib are the grey, red, green and blue intensities, respectively. - In order to categorize the pixels, develop an ImageJ plugin applying the following rules: (a) If Igr is ≤ 64, use the dark orange yeast (strain 1, Supplementary Table 3) for this pixel. (b) If 64 < Igr ≤ 128, use the orange yeast (strain 2, Supplementary Table 3) for this pixel. (c) If 128 < Igr ≤ 192, use the yellow yeast (strain 3, Supplementary Table 3) for this pixel. (d) If Igr > 192, use the white yeast (CEN.PK113-7D) for this pixel.
- Spot yeast cells to create the yeast pixel art.
- Inoculate 500 mL shake flasks containing 100 mL of YEPD (2% glucose) medium with three differently colored carotenoid producing S. cerevisiae strain and wild type CEN.PK113-7D. Incubate cultures overnight at 30 °C with shaking at 250 rpm.
- Transfer 0.5 mL of the overnight culture to a tube filled with 0.5 mL of sterile non-ionic density gradient medium (see the Table of Materials). Mix by vortexing briefly.
- Transfer the cell suspension to a qualified reservoir, 2 x 3 well. Perform spotting using an acoustic liquid handler instrument from a qualified reservoir source plate to a microplate (see the Table of Materials) containing 50 mL of YEPD (2% glucose) agar. To simplify plating, define wells on plate, e.g. use a microplate as a 6144 well plate (64 × 96).
- Spot 25 nL of each S. cerevisiae strain from the 2x 3 well reservoir source plate using a .csv file with the fluid calibration setting 6RES_AQ_GPSA2 onto the destination microplate. Define each of these 25 nL droplets as a pixel in the 64 x 96 grid which is translated to the well positions (A01, B01, C01 etc.).
- Incubate the microplate at 30 °C for 48 hours. To intensify the colors of the strains store the agar plate at 4 °C for at least 72 hours.
The protocol for multiplex genome editing using CRISRP/Cas12a was demonstrated by constructing three carotenoid producing S. cerevisiae strains expressing the crtE, crtYB and crtI genes using heterologous promoters of high, medium and low strength: strain 1, 2 and, 3 respectively (Supplementary Table 3). Construction of these strains required generation of three donor DNA expression cassettes and six flanking regions per strain for targeting to three different loci in genomic DNA (shown in Figure 2B). As described herein, promoter, open reading frame, terminator and two contiguous 50-bp connectors sequences were assembled into an expression cassette via a Golden Gate Cloning reaction and the assembly was verified by PCR (Figure 3A). The single crRNA array was ordered as a synthetic DNA fragment and was amplified by PCR (Figure 3B). The recipient plasmid for the single crRNA array (plasmid pRN1120) was linearized with EcoRI-HF and XhoI and linearization was confirmed by electrophoresis (Figure 3C). The design and nucleotide sequences of the introduced donor DNA expression cassettes and flanking regions are shown in Supplementary Table 3 and Supplementary Table 4. The sequence of single crRNA array expression cassettes is provided in Supplementary Table 1. Functionality of the spacers included in the single crRNA array was tested beforehand by singleplex genome editing with individual crRNAs19.
The efficiency of genome editing using Cas12a was firstly evaluated based on the number of colored colonies obtained after transformation (Table 1, Figure 4). The editing efficiency of the three constructed strains varied from 50% to 94%. Notably, introduction of expression cassettes used to generate strain 1 displayed the lowest editing efficiency, possibly caused by the nature of the donor DNA (i.e., these expression cassettes encode crtE, crtYB and crtI from three high strength promoters). Secondly, correct integration of the three donor DNA expression cassettes at the intended loci on the genomic DNA was confirmed by PCR (Figure 5). Primers were designed in such a way that PCR products were obtained when correct integration of donor DNA at the intended locus occurred. For each transformation experiment, eight colonies were picked from the transformation plate and tested (note that only three are presented in Figure 5). In general, out of 8 colonies tested per donor DNA, correct integration of the crtE donor DNA at the INT1 locus, crtYB at the INT2 locus and crtI at the INT3 locus was confirmed in >90% of the transformants. These results demonstrate the CRISPR/Cas12a system in combination with a single crRNA array enables efficient multiplex editing of the S. cerevisiae genome at multiple loci simultaneously.
Additionally, we demonstrate the creation of “yeast pixel art” using the three carotenoid producing strains that were constructed together with a non-colored wild-type strain. Starting from a black and white picture of Rosalind Franklin (Figure 6A), a 4-color picture (Figure 6B) and spotting list was created which was then used to spot the four different yeast strains on an agar microplate using an acoustic liquid handler, resulting in a high-resolution “yeast painting” of Rosalind Franklin (Figure 6C,D,E).
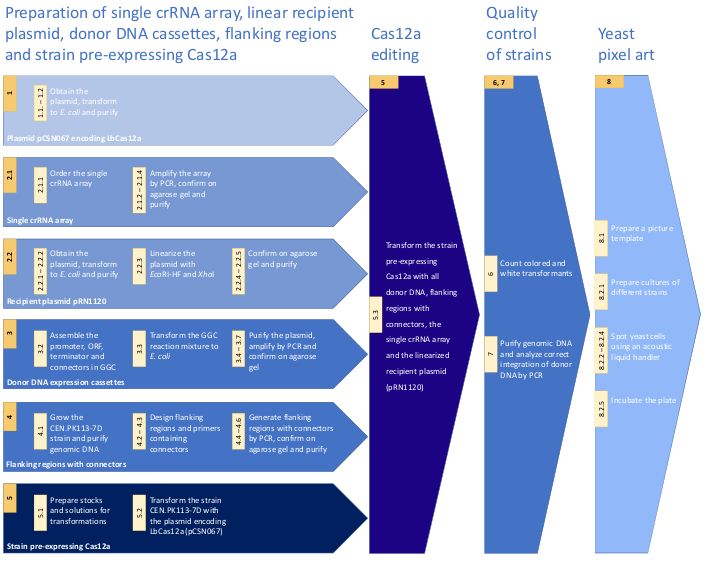
Figure 1: Workflow of the protocol for CRISPR/Cas12a multiplex genome editing in S. cerevisiae. The workflow includes crucial steps of the presented method. For details see the Protocol. Please click here to view a larger version of this figure.
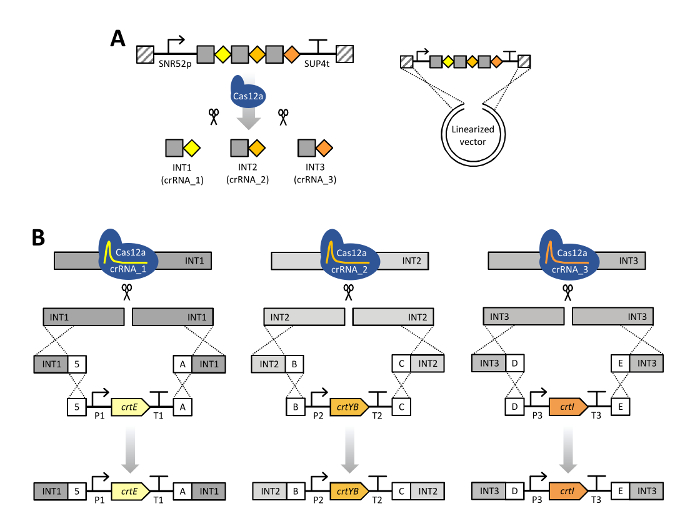
Figure 2: Scheme of CRISPR/Cas12a multiplex genome editing using a single crRNA array. (A) The single crRNA array is composed of three crRNAs units in their mature form, a 20-bp direct repeat specific for LbCas12a (grey squares) with a 23-bp guide sequence (colored diamonds). Expression of the crRNA array is enabled by the SNR52 promoter and SUP4 terminator. Transformation of S. cerevisiae with a linearized pRN1120 and the single crRNA array expression cassette containing homology with pRN1120 (diagonal stripes) allows for in vivo recombination into a circular plasmid in cells pre-expressing LbCas12a. The single crRNA array is subsequently processed by Cas12a. (B) Cas12a is directed to the intended INT1, INT2 and INT3 genomic target sites and creates double stranded breaks. In the transformation mixture, donor DNA consisting of flanking regions and the carotenoid gene expression cassette were included. Donor DNA assemblies were targeted to one stretch of DNA in genomic DNA around the INT1 (crtE), INT2 (crtYB) and INT3 (crtI) loci by in vivo recombination due to the presence of 50-bp homologous connectors sequences, indicated as 5, A, B, C, D or E. P1–P3, different promoters; T1–T3, different terminators. This figure has been modified from Verwaal et al. 201819. Genetic constructs shown using Synthetic Biology Open Language (SBOL) Visual symbols40. Please click here to view a larger version of this figure.
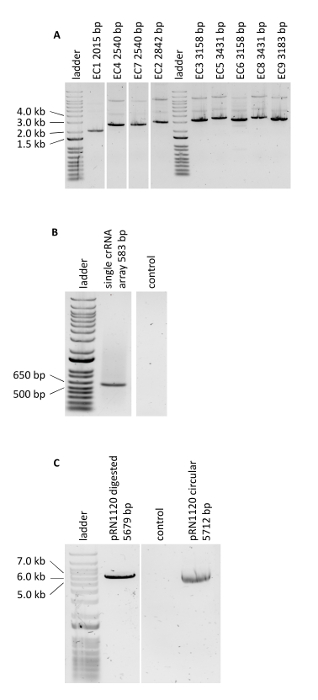
Figure 3: PCR verifying the genome editing experiments. (A) Verification of Golden Gate Cloning reactions of assembled donor DNA cassettes. Obtained results are in agreement with expected lengths. (B) PCR of the single crRNA array. (C) Linearization of plasmid pRN1120. Please click here to view a larger version of this figure.

Figure 4: Plates of S. cerevisiae transformations using the multiplex genome editing approach. (A) Strain 1 expressing crtE, crtYB and crtI from three strong promoters (dark orange colonies). (B) Strain 2 expressing crtE, crtYB and crtI from three medium strength promoters (orange colonies). (C) Strain 3 expressing crtE, crtYB and crtI from three low strength promoters (yellow colonies). Please click here to view a larger version of this figure.
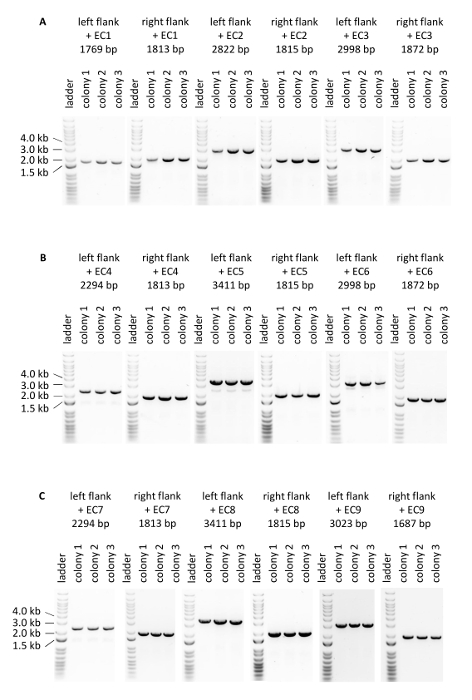
Figure 5: PCR verifying integration of the donor DNA expression cassettes at the intended loci within the genomic DNA. (A) Verification of three colonies of the strain 1. (B) Verification of three colonies of the strain 2. (C) Verification of three colonies of the strain 3. Please click here to view a larger version of this figure.

Figure 6: Yeast pixel art of Rosalind Franklin. (A) Black and white RGB photo of 220 × 280 pixels of Rosalind Franklin that was used as a template. (B) Computer conversion of the black and white photo of Rosalind Franklin into a 4-color 64 × 96 pixel list. (C) Photo of yeast pixel art with 64 × 96 yeast colonies with a zoomed-in section. (D) Photo of an acoustic liquid handler with two full grown plates. (E) Photo of a full grown microplate with 64 × 96 yeast colonies. Please click here to view a larger version of this figure.
| Strain 1 | Strain 2 | Strain 3 | |
| Colored colonies | 16 | 279 | 220 |
| White colonies | 16 | 18 | 18 |
| Total colonies | 32 | 297 | 238 |
| Efficiency | 50% | 94% | 92% |
Table 1: Editing efficiency of the multiplex genome editing approach.
| crRNA array sequencea,b,c,d,e,f |
| CATGTTTGACAGCTTATCATCGATAATCCGGAGCTAGCATGCGGCCGCTCTAGAACTAGTGGATCCCCCGGGCTGCAGTCTTTGAAAA GATAATGTATGATTATGCTTTCACTCATATTTATACAGAAACTTGATGTTTTCTTTCGAGTATATACAAGG TGATTACATGTACGTTTGAAGTACAACTCTAGATTTTGTAGTGCCCTCTTGGGCTAGCGGTAAAGGTGCGCA TTTTTTCACACCCTACAATGTTCTGTTCAAAAGATTTTGGTCAAACGCTGTAGAAGTGAAAGTTGGTGCGC ATGTTTCGGCGTTCGAAACTTCTCCGCAGTGAAAGATAAATGATCAATTTCTACTAAGTGTAGAT CTGGTGGGAGAGAAAGCTTATGAAATTTCTACTAAGTGTAGATGTGCCGTAC GCCGGAGCCGACGGAATTTCTACTAAGTGTAGATTGCCCCTCTTATACGATTATATTTT TTTTTGTTTTTTATGTCTGGGGGGCCCGGTACCCAGCTTTTGTTCCCTTTAGTGAGG GTTAATTCCGAGCTTGGCGTAATCATGGTCATAGCTGTTTCCTGTGTG |
| a. Homology to pRN1120 (bold). b. SNR52 promoter (italics). c. Genomic target sequences (underlined). d. Guide direct repeats specific for LbCas12a (italics, bold). e. SUP4 terminator (italics). f. Homology to pRN1120 (bold). |
Supplementary Table 1: Single crRNA array for LbCas12a containing homology with plasmid pRN1120.
| Name | Sequencea | Descriptionb | Used in point |
| KC-101 | CATGTTTGACAGCTTATCATC | FW primer for amplification of single crRNA array | 2.1.4 |
| KC-102 | CACACAGGAAACAGCTATGAC | RV primer for amplification of single crRNA array | 2.1.4 |
| KC-103 | AAGCGACTTCCAATCGCTTTGC | FW primer for amplification of donor DNA with connector 5 | 3.6.1 |
| KC-104 | AAAGCAAAGGAAGGAGAGAAC | RV primer for amplification of donor DNA with connector A | 3.6.1 |
| KC-105 | CGGATCGATGTACACAACCG | FW primer for amplification of donor DNA with connector B | 3.6.1 |
| KC-106 | CAACAGGAGGCGGATGGATATAC | RV primer for amplification of donor DNA with connector C | 3.6.1 |
| KC-107 | AACGTTGTCCAGGTTTGTATCC | FW primer for amplification of donor DNA with connector D | 3.6.1 |
| KC-108 | AGGTACAACAAGCACGACCG | RV primer for amplification of donor DNA with connector E | 3.6.1 |
| KC-109 | CACTATAGCAATCTGGCTATATG | FW primer for amplification of INT1 5' with connector 5 | 4.4 |
| KC-110 | AAACGCCTGTGGGTGTGGTAC TGGATATGCAAAGCGATTGGAA GTCGCTTGACTCCTCTGCCGTC ATTCC |
RV primer for amplification of INT1 5' with connector 5 | 4.4 |
| KC-111 | TTGCCCATCGAACGTACAAG TACTCCTCTGTTCTCTCCTTCCTT TGCTTTAAGCGTTGAAGTTTCCTC TTTG |
FW primer for amplification of INT1 3' with connector A | 4.4 |
| KC-112 | TGTCAACTGGAGAGCTATCG | RV primer for amplification of INT1 3' with connector A | 4.4 |
| KC-113 | AGAAGATTTCTCTTCAATCTC | FW primer for amplification of INT2 5' with connector B | 4.4 |
| KC-114 | TGCTAAGATTTGTGTTCGTT TGGGTGCAGTCGGTTGTGTACAT CGATCCGCCCTTATCAAGGATACC TGGTTG |
RV primer for amplification of INT2 5' with connector B | 4.4 |
| KC-115 | ACGCTTTCCGGCATCTTCCA GACCACAGTATATCCATCCGCCT CCTGTTGGGCGATTACACAAGCG GTGG |
FW primer for amplification of INT2 3' with connector C | 4.4 |
| KC-116 | TCTCCTCTTCGATGACCGGG | RV primer for amplification of INT2 3' with connector C | 4.4 |
| KC-117 | GGTCGTTTTTGTGCAGCATATTG | FW primer for amplification of INT3 5' with connector D | 4.4 |
| KC-118 | GCGGAATATTGGCGGAACGG ACACACGTGGATACAAACCTG GACAACGTTTTCCAAGGAGGTG AAGAACG |
RV primer for amplification of INT3 5' with connector D | 4.4 |
| KC-119 | AAATAACCACAAACATCCTT CCCATATGCTCGGTCGTGCTTGTT GTACCTGATGGGACGTCAGCACT GTAC |
FW primer for amplification of INT3 3' with connector E | 4.4 |
| KC-120 | GAGCTTACTCTATATATTCATTC | RV primer for amplification of INT3 3' with connector E | 4.4 |
| KC-121 | GTTACTAAACTGGAACTGTCCG | FW primer for verification of integration of con5-crtE-conA to INT1 5' | 7.4.1 |
| KC-122 | CACTGCTAACTACGTTTACTTC | FW primer for verification of integration of con5-crtE-conA to INT1 3' | 7.4.1 |
| KC-123 | CACTGGAACTTGAGCTTGAG | FW primer for verification of integration of conB-crtYB-conC to INT2 5' | 7.4.1 |
| KC-124 | GTCTCCAGCTGAATTGGTCC | FW primer for verification of integration of conB-crtYB-conC to INT2 3' | 7.4.1 |
| KC-125 | CTCTCATGAAGCAGTCAAGTC | FW primer for verification of integration of conD-crtI-conE to INT3 5' | 7.4.1 |
| KC-126 | GATCGGTCAATTAGGTGAAG | FW primer for verification of integration of conD-crtI-conE to INT3 3' | 7.4.1 |
| KC-127 | CCTTGTCCAAGTAGGTGTCC | RV primer for verification of integration of con5-crtE-conA to INT1 5' | 7.4.1 |
| KC-128 | GCTGTCATGATCTGTGATAAC | RV primer for verification of integration of con5-crtE-conA to INT1 3' | 7.4.1 |
| KC-129 | CTGGCAATGTTGACCAATTGC | RV primer for verification of integration of conB-crtYB-conC to INT2 5' | 7.4.1 |
| KC-130 | CCAACGTGCCTTAAAGTCTG | RV primer for verification of integration of conB-crtYB-conC to INT2 3' | 7.4.1 |
| KC-131 | CCTTACCTTCTGGAGCAGCAG | RV primer for verification of integration of conD-crtI-conE to INT3 5' | 7.4.1 |
| KC-132 | CTGGTTACTTCCCTAAGACTG | RV primer for verification of integration of conD-crtI-conE to INT3 3' | 7.4.1 |
| a. Bold sequences denote connector sequences. b. Forward and reverse primers are designated as FW and RV, respectively. |
Supplementary Table 2: Primer sequences.
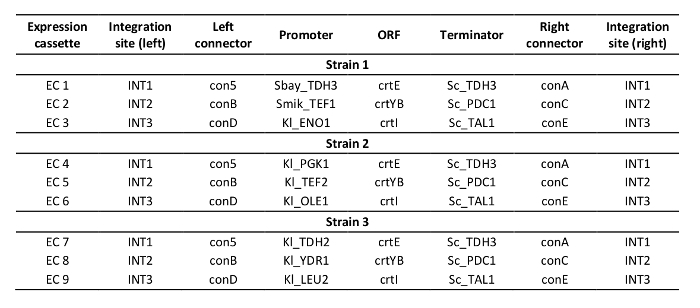
Supplementary Table 3: Design of constructed strains.

Supplementary Table 4: Sequences of donor DNA expression cassettes and flaking regions. Please click here to download this file.
