In order to analyze the effects of siRNAs/miRNAs on the cell cycle activity of postnatal cardiomyocytes in vitro, cardiomyocytes of double-transgenic Myh6-H2B-mCh/CAG-eGFP-anillin mice were isolated on postnatal day 3 (P3) and transfected with cell cycle activity-inducing miR1995, siRNA p27, and siRNA Fzr1. Compared to the negative control (Figure 1A), the pictures of miR199- (Figure 1B) and siRNA p27- (Figure 1C) transfected cardiomyocytes show an induction of cell cycle activity. In the eGFP-anillin mouse model, siRNAs against Fzr1 can be used as transfection controls, as the inhibition of Fzr1 leads to the accumulation of eGFP-anillin fusion protein in the nuclei of transfected cells and a loss of Fzr1 leads to the inhibition of APCFzr1. Fzr1 is a cofactor of the anaphase-promoting complex E3 ligase, which targets anillin for proteasomal degradation. Figure 1D shows a confocal overview picture of siRNA Fzr1-transfected cardiomyocytes 3 days after transfection, indicating a transfection efficiency of ~ 45%. Cardiomyocytes that perform endoreduplication (e.g., after knockdown of p276) show exclusively nuclear eGFP-anillin expression (Figure 1E) or are eGFP-anillin negative (see discussion). They do not express eGFP-anillin in M-phase-typical localizations (Figure 1F and G), as endoreduplication only consists of an endo-S phase (eGFP-anillin-positive) and an endo-G phase (eGFP-anillin-negative). In the endo-G phase, the APC is active, resulting in the ubiquitination and degradation of eGFP-anillin in the proteasome.
Quantification of mono- and binucleated cardiomyocyte portions at the adult stage can be performed either at the single-cell level after the Langendorff dissociation of Myh6-H2B-mCh transgenic hearts or in thick cryoslices of transgenic hearts. After the enzymatic digestion of the heart tissue at the Langendorff apparatus, atria and ventricles can be mechanically separated and analyzed independently of each other. Figure 2A shows a representative picture of non-fixed H2B-mCh transgenic ventricular cardiomyocytes after Langendorff isolation with a high degree of binucleated cardiomyocytes, as indicated by the nuclear H2B-mCh expression. By contrast, the majority of atrial cardiomyocytes are mononucleated (Figure 2B). As the enzymatic digestion does not result in 100% single cells, the pattern of cross-striation revealed by α-actinin staining facilitates discrimination between binucleated cardiomyocytes (continuous pattern of cross-striation, Figure 2C) and cell doublets. Figure 2D shows an example of the identification of a binucleated cardiomyocyte in a thick slice.
3D reconstructions of thick slices of adult Myh6-H2B-mCh transgenic hearts can be used to determine the proportion of cardiomyocyte nuclei under physiological conditions within the tissue. Using the 3D module of imaging software, Hoechst-stained nuclei and H2B-mCh-positive nuclei can be detected and counted automatically, as illustrated in Figure 2E. The final result should be corrected manually for doublets, meaning nuclei that directly touch each other, in this case. To analyze the nucleation index of cardiomyocytes in thick slices, it is necessary to manually scroll through the stack, as the nuclei do not necessary lie within one z-plane. WGA staining allows for the detection of cell borders.
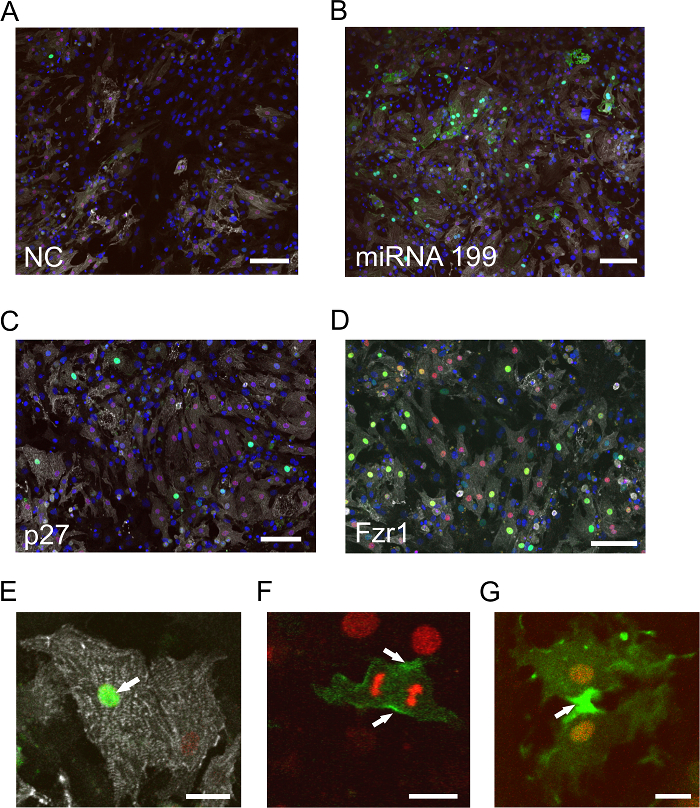
Figure 1: Examples of In Vitro Visualization of Cell Cycle Activity in Postnatal Cardiomyocytes after Transfection with siRNAs. (A-D) P3 cardiomyocytes from eGFP-anillin/Myh6-H2B-mCh hearts stained for α-actinin (white). Cardiomyocyte nuclei are identified by the H2B-mCh signal (red), cell cycle activity by eGFP-anillin signals (green), and nuclei by Hoechst nuclear dye (blue). (A) P3 cardiomyocytes transfected with scramble siRNA serve as the negative control. The bar is 100 µm. (B) P3 cardiomyocytes transfected with miRNA-199 display significantly more eGFP-anillin signals than the control (A). The bar is 100 µm. (C) Example of exclusively nuclear eGFP-anillin signals after transfection with p27 siRNAs, indicating endoreduplication. The bar is 80 µm. (D) Transfection with siRNA against Fzr1 for the determination of transfection efficiency. As eGFP-anillin accumulates, the number of eGFP-anillin+ cardiomyocytes indicates the transfection efficiency. The bar is 80 µm. (E-G) Different localizations of eGFP-anillin (green) in cardiomyocytes during the cell cycle: nuclear localization (arrow in E), contractile ring (arrows in F), and midbody localization (arrow in G). The bar is 20 µm in (E) and 10 µm in (F and G). Please click here to view a larger version of this figure.

Figure 2: Examples for the Assessment of Multinucleation by the Langendorff Isolation of Cardiomyocytes from H2B-mCh Mice and by the 3D Analysis of Thick Slices. (A,B) Ventricular (A) and atrial (B) cardiomyocytes from hearts from adult H2B-mCh transgenic mice after isolation by Langendorff dissociation. The bars are 50 µm. (C) Cardiomyocytes from Myh6-H2B-mCh hearts stained for α-actinin (green). Cardiomyocyte nuclei are identified by the H2B-mCh signal (red). The bar is 10 µm. (D) Binucleated cardiomyocyte in a thick slice (arrows). Cardiomyocyte nuclei are identified by the H2B-mCh signal (red), cell borders by WGA staining (green), and nuclei by ToPro3 (white). The bar is 50 µm. (E) Workflow for the 3D analysis of binucleation in thick slices. Please click here to view a larger version of this figure.
