During the separation and purification steps, samples for SDS-PAGE electrophoresis were taken and subsequently analyzed using Coomassie-blue staining (Figure 2). 20 µL of each sample was mixed with 10 µL of Laemmli sample buffer (188 mM Tris-HCl pH 6.8, 3% SDS (w/w), 30% glycerol (v/v), 0.01 bromophenol blue (w/w), 15% β-mercaptoethanol) and incubated at 95 °C for 15 min. 4 µL of each sample was loaded on 12.5% acrylamide SDS gel and separated under a constant current of 30 mA per gel in reducing and denaturing conditions.
The results confirmed an incremental increase in the relative tubulin concentration accompanied by a reduction in contaminating proteins. Moreover, there was no significant loss of tubulin in clarified lysate in the first centrifugation (step 3.2.7) compared to omitting this step (Figure 2A,B).
The protein concentration was determined using three independent methods: BCA assay, Bradford protein assay, and SDS-PAGE gel densitometry analysis19 (Figure 3). The overall yield of tubulin using the procedure described was 123 mg of purified tubulin from 250 g of neural tissue. During the measurements, it is necessary to take into account that the high DTT concentration in the storage buffer has a significant impact on the BCA assay. Both the PEM buffer and PBS buffer, with the addition of DTT, increase the background absorbance by about 0.900 A595, which significantly reduces the capacity of the BCA assay (Figure 3A). The negative effect of DTT is detectable even after ten times dilution with pure water (data not shown). The Bradford assay did not seem to be affected by the storage buffer (Figure 3B), as confirmed by densitometry analysis.
The purity of tubulin preparation was verified by mass spectrometry analysis at two independent facilities (VRI Brno, Czech Republic; CEITEC MU Brno, Czech Republic) using electrospray ionization and MALDI-TOF. Both analyses confirmed the presence of porcine tubulins alpha and beta in several isoforms. The overall purity was more than 97.07% (PSMs 1065), with the most prevalent impurities originating from Homo sapiens Keratin Type II (PSMs 246, 2.24% of impurities) which were introduced most probably during the tubulin isolation and sample preparation for MS/MS analysis. Other impurities composed of 322 PSMs and only Serum albumin, Actin gamma, and Trypsinogen originating from Sus scrofa were identified with one peptide resolution (0.0069%).
In subsequent experiments, the preservation of polymerization capacity after snap freezing and storage in liquid nitrogen was verified. The 10 mg/mL aliquot was removed from liquid nitrogen and slowly thawed on ice. Samples of different concentrations (10 mg/mL, 8 mg/mL, 6 mg/mL, 4 mg/mL, and 2 mg/mL) were prepared by diluting the aliquots in PEM buffer containing DTT, ATP, and GTP for the self-assembly experiment. One dilution series was incubated at 37 °C for 60 min, and the second one was incubated on ice for 60 min. Both series were centrifuged for 60 min at 21,000 x g and the corresponding temperature (4 °C or 37 °C). 30 µL of supernatant was removed and used for SDS-PAGE. The remaining supernatant was carefully removed using a pipette and discarded. Pellets were briefly washed by adding 100 µM of PEM buffer, followed by immediate removal using a pipette. Subsequently, the pellets were resuspended in 50 µL of 1x concentrated SDS-loading buffer so that the relative concentration of pellet and supernatant is preserved. 10 µL of SDS-loading buffer was added to each supernatant. All samples were analyzed using the SDS-PAGE and Coomassie Staining (Figure 4). The volume of each sample loaded to SDS-PAGE was adjusted according to the starting concentration, so the difference in precipitation due to the concentration is more obvious. The self-assembly test of tubulin in PEM buffer confirmed the ability to form tubulin fibers in a temperature-dependent manner.
The MAP2c driven tubulin20,21 assembly assay verifying the ability of tubulin to interact with other proteins was performed22. The serial dilutions of MAP2c were prepared where 100 µL of 1 mg/mL tubulin aliquots were mixed with MAP2c to the final concentration ranging from 0 µM to 8 µM. All samples were prepared on ice by diluting in freshly prepared PEM buffer with 1 mM DTT and 1 mM GTP. The tubulin was incubated for 15 min at 37 °C with different concentrations of MAP2c, and centrifuged for 60 min at 21,000 x g and 37 °C. The final wash of the pellet was performed twice with 100 µL of PEM buffer. The experiment confirmed the capacity of the prepared tubulin to undergo MAP2c-driven polymerization, as only the sample without MAP2c did not form a pellet (Figure 5).
Transmission electron microscopy was further used to confirm the presence of tubulin filaments from co-polymerization experiments. Suspensions of purified tubulin precipitated with MAP2c were prepared for transmission electron microscopy using negative staining. The samples were adsorbed onto Formvar-coated, carbon-stabilized copper grids. The grids were then negatively stained with 2% NH4MoO4 and examined under an electron microscope at 18,000x magnification and an accelerating voltage of 80 kV. The presence of homogenous microtubules with a clear filamentous structure and appropriate size showed the ability to form microtubules in native conformation (Figure 6).
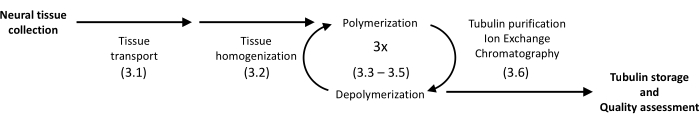
Figure 1: A schematic diagram of tubulin separation and purification. The number in parentheses refers to the protocol step. Please click here to view a larger version of this figure.
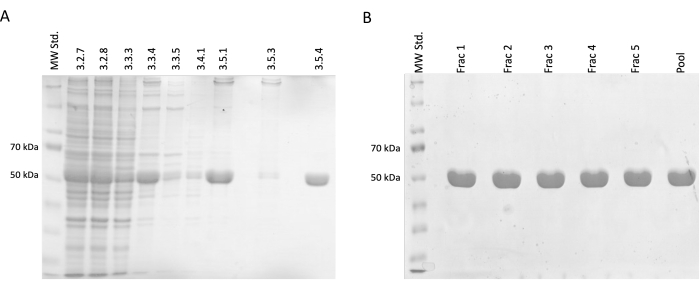
Figure 2: Tubulin separation and purification. (A) The SDS-PAGE analysis of samples taken during the separation of tubulin utilizing temperature-driven polymerization (3 µL per line). The decrease in impurities with a stable increase in relative tubulin abundance (approximately 50 kDa) is visible. The numbers above each line correspond to the step number in the protocol. (B) The SDS-PAGE analysis of fractions obtained after protein chromatography on phospho-cellulose resin (4 µL per line). Please click here to view a larger version of this figure.
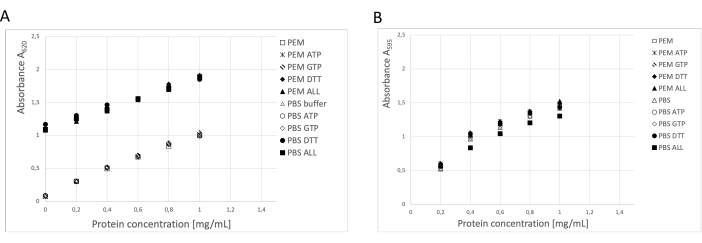
Figure 3: Protein concentration. The dilution series of bovine serum albumin of up to 1 mg/mL in PEM buffer or PBS was compared with BSA dilution series containing 0.1 ATP, 1 mM GTP, 1 mM DTT separately or in combination (0.1 mM ATP, 1 mM GTP, and 1 mM DTT) in the PEM or PBS buffer. (A) When the BCA assay was used, there was a significant shift in the background of samples containing DTT (solid symbols). (B) This effect was not detected when concentrations were measured in the Bradford assay. Please click here to view a larger version of this figure.
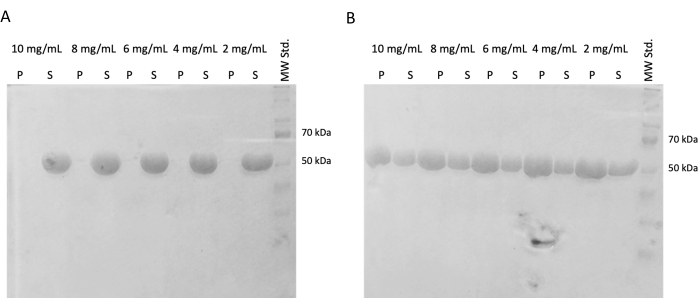
Figure 4: Tubulin self-assembly assay. The SDS-PAGE analysis of the self-assembly assay confirmed the capacity of stored tubulin incubated at either (A) 4 °C or (B) 37 °C to polymerize in a temperature-dependent manner in a broad spectrum of concentrations. (P – pellet, S – supernatant; the respective concentration of tubulin is denoted above each line pair; the amount of each sample loaded was 10 µg). Please click here to view a larger version of this figure.
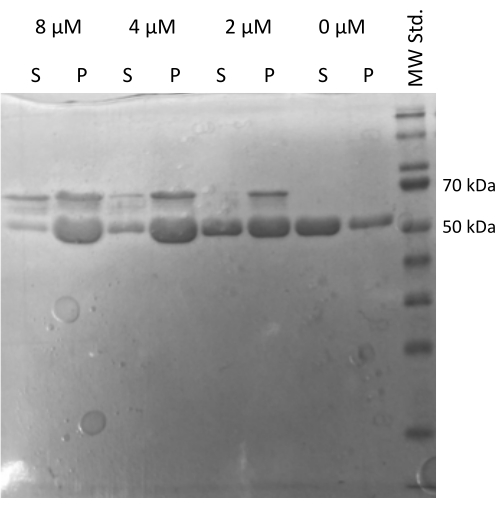
Figure 5: Tubulin co-sedimentation assay. The SDS-PAGE analysis of MAP2c-assisted tubulin assembly confirmed the capacity of stored tubulin to undergo polymerization driven by interaction with microtubule-associated protein in a concentration-dependent manner. The molar concentration of MAP2c is indicated above each line. (P – pellet, S – supernatant). Please click here to view a larger version of this figure.
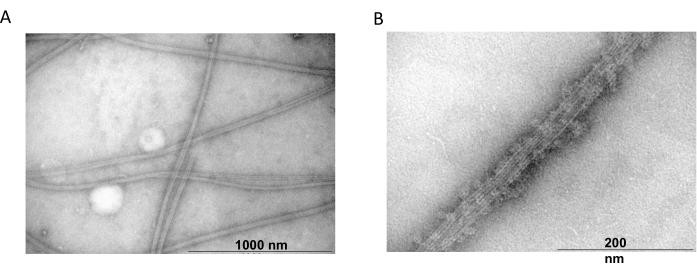
Figure 6: Transmission electron microscopy. The TEM microphotographs showed that tubulin assembles (A) into microtubules of appropriate diameter (B) consisting of homogenous tubulin filaments decorated with MAP2c proteins. The bar corresponds to 1000 nm (A) and 200 nm (B). Please click here to view a larger version of this figure.
