1. Reagents and buffers
NOTE: All sterilization using vacuum filters should be performed with 0.2 µm polyethersulfone (PES) membrane in a biosafety cabinet.
- Prepare RNase-free water by adding 1.0 mL of diethylpyrocarbonate (DEPC) to 1.0 L of deionized water, mix for at least 1 hr at room temperature (RT), autoclave twice, and then cool to RT before use.
- Prepare Dulbecco's Modified Eagle Medium (DMEM) by mixing one packet of DMEM powder (13.4 g), 3.7 g of sodium bicarbonate, 100 mL of fetal bovine serum (FBS), penicillin, and streptomycin to ~800 mL of sterile deionized water. The final concentration of penicillin and streptomycin in DMEM should be 50 U/mL and 50 µg/mL, respectively. Adjust the pH to 7.4 and then make up the volume to 1.0 L with sterile deionized water. Sterilize by filtration and store at 4 °C.
- Prepare 10x phosphate buffered saline (10x PBS) by adding 25.6 g of disodium hydrogen heptahydrate (Na2HPO4·7H2O), 2 g of potassium dihydrogen phosphate (KH2PO4), 2 g of potassium chloride (KCl), and 80 g of sodium chloride (NaCl) to 800 mL of deionized water. Mix to dissolve and make up the volume to 1.0 L. Sterilize by filtration and store at 4 °C.
- Prepare 0.5 M ethylene diamine tetra-acetic acid (EDTA) by dissolving 186.1 g of Na2•EDTA•2H2O into ~800 mL of deionized water. Adjust pH to 8.0 and then make up the volume to 1.0 L with sterile deionized water. Sterilize by filtration and store at 4 °C.
- Prepare 1x trypsin-EDTA solution by mixing 100 mL of 10x trypsin (2.5%), 2 mL of 0.5 M EDTA, and add 1x PBS up to 1.0 L. Sterilize by filtration and aliquot into 50 mL conical tubes. Store at 4 °C for 1-2 weeks or freeze at -20 °C for long-term use.
- Prepare 2x formamide DNA/RNA loading dye by mixing 14.4 mL of formamide and 0.6 mL of 0.5 M EDTA for a final volume of 15 mL. Add bromophenol blue and xylene cyanol powder to a final concentration of 0.02% and store at 4 °C.
NOTE: Formamide is toxic and corrosive. Read the material safety data sheets for additional safety recommendations. - Prepare 5x Tris/Borate/EDTA (5x TBE) buffer by mixing 54.0 g of tris base, 27.5 g of boric acid, and 20 mL of 0.5 M EDTA into ~800 mL of deionized water. Mix to dissolve and make up the volume to 1.0 L with deionized water.
- Prepare urea-polyacrylamide gel electrophoresis (urea-PAGE) solution by mixing 200 mL of 5x TBE, 250 mL of 40% 19:1 bis/acrylamide, and 450.5 g of urea. Then add deionized water up to 1.0 L. Mix until the ingredients are completely dissolved, then sterilize by filtration, and store at 4 °C in an amber glass bottle.
NOTE: Bis/acrylamide is toxic. Read the material safety data sheets for additional safety procedures. - Prepare 10% ammonium persulfate (APS) by dissolving 1 g of APS in 10 mL of deionized water and store at 4 °C.
2. Cotransfection of HeLa cells with the reporter and U1 snRNA plasmids
NOTE: The transfection of Hela cells must be performed under sterile conditions in a biological safety cabinet. The outer surface of all materials must be sprayed with 70% ethanol before being introduced into the biological safety cabinet.
- Maintain Hela cells in DMEM in a 37 °C incubator with 5% CO2 by passaging every 2-3 days when the cells are about 80-90% confluent.
- For passaging HeLa cells, aspirate the spent medium and then incubate cells with 3 mL of 0.25% trypsin containing 1 mM EDTA at 37 °C for 3 min.
- After incubation, add 7 mL of fresh DMEM. Transfer the cell suspension to a 10 mL tube, centrifuge at 1,000 x g for 5 min.
- Resuspend the cell pellet in 10 mL of fresh DMEM, and then plate the cells on a new tissue culture dish at 20% confluence.
- For transient transfections, count Hela cells with a clean hemocytometer slide and prepare a suspension with a density of 2.5 x 105 cells/mL.
- Seed 1.0 mL of 2.5 x 105 cells/mL Hela cell suspension into each well of a 12-well plate and incubate overnight at 37 °C.
- The next day, aspirate the spent medium and add 0.8 mL of fresh DMEM with serum.
- Prepare Solution I by adding 0.2 µg of Dup51 or Dup51p reporter plasmid, 1.8 µg of pcDNA, pNS6U1, or pNS6U1-5a plasmid, and 100 µL of transfection medium into a new 1.5 mL microcentrifuge tube.
- Prepare a master mix of Solution II for all samples to be transfected by adding 100 µL of transfection medium and 4.0 µL of transfection reagent per sample.
- Prepare the transfection mix by adding 100 µL of Solution II into each microcentrifuge tube containing Solution I.
- Vortex the transfection mixes for 15 s, centrifuge in a tabletop microcentrifuge at 3,000 x g for 10 s at RT, and then incubate at RT for 5 min.
- Add all 200 µL of the transfection mix into one well of the 12-well HeLa cell plate to achieve a final volume of 1.0 mL per well and incubate the plate at 37 °C for 48 hrs.
- After incubation, extract RNA from the transfected HeLa cells with commercially available guanidine thiocyanate and phenol solution.
NOTE: This reagent contains phenol and this step should be performed in a fume hood. The use of DEPC treated water is recommended for resuspension of extracted RNA.- Aspirate the spent medium and add 500 µL of the reagent into each well. Incubate at RT for 5 min.
- Homogenize by pipetting up and down. Then transfer the solution to a new 1.5 mL microcentrifuge tube.
- Add 100 µL of chloroform and vortex for 15 s.
- Centrifuge at 12,000 x g for 15 min at RT.
- Transfer 200 µL of the RNA containing, top aqueous layer to a new 1.5 mL microcentrifuge tube.
- Add 2 µg of glycogen and 200 µL of isopropanol to each RNA sample. Mix by inverting the tubes.
- Collect the RNA precipitate by centrifugation at 12,000 x g for 10 min at 4 °C.
- Remove and discard the supernatant without disturbing the RNA pellet.
- Wash the pellet twice by adding 1.0 mL of 70% ethanol, inverting the tubes, and centrifuging as described in Step 2.13.7.
- Air dry the pellet for ~10-20 min at RT and resuspend the RNA in 10-20 µL of RNase-free water.
- Determine RNA concentration by measuring absorbance at 260 nm as described by Desjardins and Conklin9.
- Proceed with primer extension or store RNA samples in -20 °C. Isolated RNA can be stored at -20 °C for 6-12 months.
3. 32P-labeling of oligonucleotides
NOTE: Steps involving the use of 32P-ATP and 32P-labeled oligonucleotides must be performed behind an acrylic shield by trained individuals with approval from authorized institutional entities. The protocol described below can be used for labeling of oligonucleotides, Dup3r and U17-26-R, and markers for urea-PAGE. Use of DEPC treated water is recommended for resuspension of oligonucleotides and size exclusion beads.
- To a 1.5 mL microcentrifuge tube, add oligonucleotide, T4 polynucleotide kinase (T4 PNK), T4 PNK buffer, and 32P-ATP as described in Table 1. Add 32P-ATP last to the mixture; this is important.
NOTE: For the addition of radioactive solutions, use of filter tips is recommended. - Incubate in a water bath at 37 °C for 30 min.
- While the labeling reactions are incubating, resuspend the size exclusion beads with a 25 kDa molecular weight cut off by gently vortexing for ~10 s.
NOTE: The size exclusion beads should be prepared according to the manufacturer's instructions and stored as a 50% suspension in 25% ethanol at 4 °C. - Prepare columns by transferring 600 μL of the resuspended beads to a disposable mini column placed in a 1.5 mL collection tube and returning the bead stock to 4 °C.
- Centrifuge at 2,000 x g for 1 min at RT and discard the flow through.
- Wash the beads by adding 300 µL of RNase-free water to the column.
- Repeat steps 3.5. and 3.6. twice and transfer the mini-column to a new 1.5 mL centrifuge tube.
- Add the kinase reaction mix to the size-exclusion bead column and centrifuge at 5000 x g for 1 min at RT.
- Collect and save the flow through, which has the 32P-labeled oligonucleotide, and discard all tips and tubes into an acrylic waste box.
- Add 20 µL of RNase-free water to dilute 32P-labeled oligonucleotide to a final concentration of 2.5 µM.
NOTE: Dilute the labeled markers as needed for loading on urea-PAGE gels. - Store the labeled oligonucleotide in an acrylic microtube box at -20 °C or proceed with primer extension analysis.
4. Analysis of the spliced reporter transcripts by primer extension
NOTE: It is recommended to clean surfaces and equipment with an RNase inactivating reagent before use.
- Add 2.0 µg of RNA extracted from the Hela cells into separate 200 μL microcentrifuge tubes and add RNase-free water to make up the volume to 6.55 µL.
- Prepare Master Mix I with the diluted 32P-Dup3r and dNTPs as shown in Table 2 and add 0.9 µL of the mix to each PCR tube containing RNA samples.
- Incubate the tubes, first at 65 °C for 5 min and then on ice for 1 min.
- Prepare Master Mix II with 5x First Strand Buffer, dithiothreitol (DTT), RNase inhibitor, and reverse transcriptase as shown in Table 2.
- Add 2.55 µL of the mix to each tube containing RNA and Master Mix 1; the total volume of the reaction should be 10 µL. Keep the tubes at RT for 10 min.
- Transfer the tubes to a dry bath or a thermal cycler and incubate, first at 50 °C for 60 min and then at 70 °C for 15 min.
- After incubation, add 10 µL of 2x formamide RNA loading dye to each sample and store in an acrylic box at -20 °C or proceed with the separation of fragments by urea-PAGE using a 14-cm long gel and visualization of gel image as described below in Step 8.
- Perform densitometric scanning of the gel image with an image analysis software and use the band intensities of included and skipped products to calculate the percentage of exon 2 inclusion as shown below.
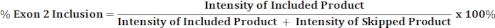
5. Analysis of the spliced reporter transcripts by fluorescent RT-PCR
NOTE: The RT-PCR analysis described below uses random hexamers for cDNA synthesis and a combination of unlabeled Dup8f and Cy5-labeled Dup3r oligonucleotides for PCR amplification of the spliced products.
- For cDNA synthesis, add 2.0 µg of RNA extracted from the transfected Hela cells into separate 200 μL microcentrifuge tubes and add RNase-free water to make up the volume to 11.0 µL.
- Prepare Master Mix I, containing random hexamers and dNTP as shown in Table 3 and add 2.0 μL of the mix to each sample. Incubate, first at 65 °C for 5 min then on ice for 1 min.
- Prepare Master Mix II, containing First Strand Buffer, RNase inhibitor, DTT, and reverse transcriptase as shown in Table 3 and add 7.0 μL of the mix to each tube containing RNA and Master Mix I.
- Keep the tubes at RT for 10 min, and then incubate at 50 °C for 60 min and 70 °C for 15 min.
NOTE: Completed cDNA reactions may be stored at -20 °C. - For PCR, transfer 1.0 μL (100 ng/μL) of each cDNA sample to new PCR tubes.
- Prepare a master mix consisting of Dup8f, Cy5-Dup3r, dNTPs, Taq buffer, Taq polymerase, and water as described in Table 4 and add 11.5 μL of the mix to each tube containing cDNA.
- Perform PCR using a thermal cycler with an initial denaturation step at 94 °C for 3 min; followed by 20 cycles of denaturation (94 °C for 30 s), annealing (65 °C for 30 s), and extension (72 °C for 15 s), and a termination step at 72 °C for 5 min.
- Add 12.5 µL of 2x formamide DNA loading dye to each tube and heat at 95 °C for 5 min.
- Store the PCR reaction at -20 °C or proceed with urea-PAGE as described below in Step 8.1-8.4.
- After electrophoresis, remove the glass plates from the electrophoresis apparatus and scan using a fluorescence imager to visualize the gel.
- Perform densitometric scanning of the gel image and use band intensity of the included and the skipped products to calculate percentage of exon 2 inclusion as described in Step 4.8.
6. Analysis of variant U1 snRNA expression by primer extension
- Add 2.0 µg of RNA extracted from the Hela cells into separate 200 μL microcentrifuge tubes and add RNase-free water to make up the volume to 4.325 µL and then add dATP, as shown in Table 5.
- Add 10,000 CPM of the 32P-U17-26-R oligonucleotide to each tube.
NOTE: To prepare a 10,000 cpm/μL solution of 32P-U17-26-R (from Step 3), dilute the labeled oligonucleotide (1:20 dilution), determine cpm in 1.0 μL using a scintillation counter, and further dilute with deionized water to prepare a solution of 10,000 cpm/μL in a new 1.5 mL microcentrifuge tube. - Incubate at 65 °C for 5 min, and then on ice for 1 min.
- Prepare a master mix with 5x First Strand Buffer, RNase inhibitor, DTT, and reverse transcriptase as shown in Table 5 and add 1.8 µL of the mix to each sample.
- Incubate, first at RT for 10 min and then at 42 °C for 10 min.
- After incubation, add 10 µL of 2x formamide RNA loading dye into each sample and store in an acrylic box at -20 °C or proceed with separation of fragments by urea-PAGE using a 38-cm long gel (see Step 8).
7. Analysis of variant U1 snRNA expression by RT-qPCR
- Dilute the cDNA stock prepared as described above in Steps 5.1 – 5.4 to a concentration of 0.2 ng/µL.
- Pipette 5.0 µL of diluted cDNA into individual wells of a 96-well qPCR plate in triplicate. Add deionized water instead of cDNA for no-template control (NTC).
- Prepare two separate primer mixes consisting of the forward and reverse primers for U1 and U2 snRNAs, and water as shown in Table 6.
NOTE: Sequences for U1 and U2 snRNA specific primers are provided in Table 7. - Add 5.0 µL of the U1 and U2 snRNA primer mix to the sample and NTC wells.
- Add 10.0 µL of real-time PCR mix to each well.
- Seal the plates with an optical film, then centrifuge at 1,000 x g for 2 min at RT to collect the reactions to the bottom of the wells.
- Perform qPCR with an initial denaturation step at 95 °C for 10 min, followed by 40 cycles of a 2-step protocol consisting of denaturation (95 °C for 15 s) and annealing/extension (62 °C for 60 s) while collecting the threshold quantification cycle (Cq) values of target amplicons.
- Finish the qPCR reaction by checking for a single peak in the dissociation curve for U1 and U2 snRNA reactions.
- From the Cq values, calculate delta Cq (ΔCq) for U1 and U2 snRNAs as compared to the pcDNA control.
- Determine variant U1 snRNA expression as relative quantity (RQ) of U1 as compared to U2 using the ΔΔCq value as shown below for all samples.
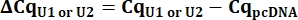


8. Setup and running of Urea-PAGE gels
NOTE: Assembly of glass plates and the gel running apparatus should be performed according to manufacturer's instructions. The casting of the 10% urea-PAGE gel can be performed according to a previously described protocol by Summer et al.10. Steps involving the preparation of markers and samples, and of running and visualization of gels are described below. Optionally, to prevent the gel from sticking to glass plates, the inner surface may be coated with silicone solution by adding 1 mL of the solution onto the surface and evenly spreading over the entire surface with tissue. Once dry, the plates should be washed with deionized water and dried again.
CAUTION: Unpolymerized acrylamide is neurotoxic and must be handled with protections recommended in the material safety data sheet.
- Prepare the primer extension samples and markers by heating at 95 °C for 5 min and then centrifuging in a tabletop microcentrifuge at 3,000 x g for 5 s at RT.
- Prior to loading the markers and samples, flush out the wells with 1x TBE buffer to remove the settled urea.
- Load 10 μL/sample/well and run the gel at 300-500V for 2-3 hrs or until the xylene cyanol reaches the bottom.
NOTE: About 1,000 cpm of the 32P-labeled marker can be loaded. - After electrophoresis, remove the glass plates from the electrophoresis apparatus.
- Carefully separate the two plates so that the gel lays flat on either glass surface and transfer the gel onto a filter paper and cover it with plastic wrap.
- Vacuum dry the gel on the filter paper at 80 °C for 30 min using a gel dryer.
- Place the dried gel in a phosphor imaging cassette and keep at RT overnight.
NOTE: The phosphor screen should be erased using a light box prior to use. - To visualize the gel image, remove the screen and scan using a phosphor imager.
The splicing reporter Dup51, a three exon-two intron minigene, was derived from the human β-globin gene and has been described previously (Figure 1A)11,12 . We created a mutant reporter, Dup51p, by introducing Usher syndrome associated 5´-splice site mutations that occur in exon 3 of the protocadherin 15 (PCDH15) gene13. The 5´-splice site sequence at the exon 2-intron 2 junction was changed from CAG/GUUGGUAUC to AUG/GUGUGUAUC (/ is the exon-intron boundary) (Figure 1B)14. These sequence changes alter the predicted pattern of 5´-splice site basepairing with the endogenous U1 snRNA and also result in skipping of exon 2 in the mature transcript (Figure 2). The effect of these mutations could be suppressed by a compensatory change in the 5´-region of the U1 snRNA. Substitution of the uracil with adenine at the 5th position of the U1 snRNA (U1-5a) reversed the effect of 5´-splice site mutations in Dup51p (Figure 1B).
The splicing of the Dup51 and Dup51p transcripts can be monitored by primer extension using 32P-Dup3r or by semi quantitative RT-PCR using Dup8f and Dup3r oligonucleotides (Figure 1); sequences are provided in Table 7. When expressed in HeLa cells, the Dup51 minigene expresses the mature full-length transcript in which exon 2 is spliced-in efficiently (Figure 2A,B, lane 1). The 5´-splice site mutations in Dup51p reduce exon 2 inclusion from ~95% to ~30%, leading to the formation of the shorter transcript (lane 5). Coexpression of Dup51p with U1-5a snRNA rescues exon 2 splicing and restores formation of full-length transcript (lane 8). Coexpression with wildtype U1 (U1-WT) or U1-5g variant does not have a similar effect on Dup51p splicing (lanes 6 and 7). U1-WT, U1-5g, or U1-5a coexpression also does not alter the splicing pattern of the Dup51 reporter (lanes 2-4). Overall, these results indicate that the rescue of exon 2 splicing in Dup51p transcripts is specifically due to the U>A mutation in the U1-5a snRNA.
To monitor the expression of transfected U1 snRNAs, primer extension or RT-qPCR can be applied. Detection of U1-5a variant transcripts by primer extension uses the U17-26 oligonucleotide, which is 20 nucleotides long and basepairs near the adenine at the 5th position of U1 snRNA (Figure 3A). In the presence of dATP alone, U17-26 allows incorporation of 2-3 nucleotides for the endogenous wildtype U1 snRNA yielding 22 or 23 nucleotides long product (Figure 3B, lanes 1, 2, 5, and 6). For the U1-5a and U1-5g variants, extension terminates after addition of a single adenosine producing a 21 nucleotides long product (lanes 3, 4, 7, and 8). Analysis by RT-qPCR allows estimation of fold increase in expression of the variant U1 over the endogenous wildtype snRNA. Here, U1 specific primer pairs are designed so that no overlap exists with the mutations introduced into the variant snRNAs (Figure 3A). After normalization to U2 snRNA expression, the transfected U1-WT, and the U1-5g and U1-5a variants were found to be expressed at 3-5 folds over the endogenous snRNA (Figure 3C). Endogenous U1 is estimated to be present at 106 copies per cell in HeLa cells15. Thus, the exogenous variant snRNAs are expressed at ~3-5 fold over the endogenous U1. The ability of the U1-5a snRNA to rescue exon 2 inclusion to ~95% demonstrates that levels of the exogenous variant snRNAs are sufficient for the splicing of the nascent reporter transcripts.
| Ingredients | Amount for One Reaction |
| PNK Buffer (10x) | 2.0 µL |
| T4 Polynucleotide Kinase (T4 PNK) (10,000 U/mL) | 1.0 µL |
| Oligonucleotide* (10 µM) | 10.0 µL |
| 32P-ATP (10 mM) | 1.0 µL |
| H2O (DEPC treated) | 6.0 μL |
| *For labeling markers, 0.5 µL of the pBR322 DNA-MspI Digest or the Low Molecular Weight Marker is used. | |
Table 1: 32P-labeling of oligonucleotides. Reaction for preparation of a 5′-32P-labeled primer using 32P-ATP.
| Ingredients – Master Mix I | Amount for One Reaction |
| RNA and H2O (DEPC treated) | 6.55 µL |
| 32P-Dup3r (2.5 µM) | 0.4 µL |
| dNTP (10 mM) | 0.5 µL |
| Ingredients – Master Mix II | Amount for One Reaction |
| First-Strand Buffer (5x) | 2.00 µL |
| RNase Inhibitor (40 U/µL) | 0.25 µL |
| DTT (0.1 M) | 0.05 µL |
| Reverse Transcriptase (RT) (200U/µL) | 0.25 µL |
Table 2: 32P-Dup3r primer extension. Reaction for the analysis of spliced reporter transcripts by primer extension.
| Ingredients – Master Mix I | Amount for One Reaction |
| RNA and ddH2O (DEPC treated) | 11.0 µL |
| Random Hexamer (50 ng/µL) | 1.0 µL |
| dNTP (10 mM) | 1.0 µL |
| Ingredients – Master Mix II | Amount for One Reaction |
| First-Strand Buffer (5x) | 4.0 µL |
| RNase Inhibitor (40 U/µL) | 1.0 µL |
| DTT (0.1 M) | 1.0 µL |
| Reverse Transcriptase (RT) (200U/µL) | 1.0 µL |
Table 3: cDNA Synthesis. Reaction for the synthesis of cDNA from total RNA.
| Ingredients | Amount for One Reaction |
| cDNA Template (100 ng/µL) | 1.00 µL |
| Forward Primer (Dup8f) (10 µM) | 0.25 µL |
| Cy5 Reverse Primer (Cy5-Dup3r) (10 µM) | 0.25 µL |
| dNTP (10 µM) | 0.25 µL |
| Taq Buffer (10x) | 1.25 µL |
| Taq DNA Polymerase (5000 U/mL) | 0.25 µL |
| H2O (DEPC treated) | 9.25 µL |
Table 4: Fluorescent RT-PCR. Reaction for the analysis of the spliced reporter transcripts by RT-PCR using Cy5-labeled primer.
| Ingredients | Amount for One Reaction |
| RNA and H2O (DEPC treated) | 4.325 µL |
| dATP (10 mM) | 0.375 µL |
| 32P-U17-26-R oligo nucleotide* | 1.000 µL |
| Ingredients – Master Mix | Amount for One Reaction |
| First-Strand Buffer (5x) | 1.5 µL |
| RNase Inhibitor (40 U/µL) | 0.1 µL |
| DTT (0.1 M) | 0.1 µL |
| Reverse Transcriptase (RT) (200U/µL) | 0.1 µL |
| *10,000 CPM of the 32P-U17-26-R oligonucleotide should be added to the diluted RNA (Step 6.2.). | |
Table 5: 32P-U1-snRNA primer extension. Reaction for the analysis of variant U1 snRNA expression by primer extension.
| Ingredients | Amount for One Reaction |
| Real Time PCR Mix | 10.0 µL |
| cDNA (0.2 ng/µL) | 5.0 µL |
| Forward Primer (10 µM) | 0.2 µL |
| Reverse Primer (10 µM) | 0.2 µL |
| H2O (DEPC treated) | 4.6 µL |
Table 6: Quantitative RT-qPCR. Reactions for quantitative analysis of variant U1 snRNA expression by RT-qPCR.
| Oligo Name | Sequence (5′ to 3′) | Technique |
| Dup8f | GACACCATGCATGGTGCACC | Fluorescent RT-PCR |
| Cy5-Dup3r | /5Cy5/AACAGCATCAGGAGTGGACAGATCCC | Fluorescent RT-PCR |
| Dup3r | AACAGCATCAGGAGTGGACAGATCCC | Primer Extension |
| U17-26R | TGGTATCTCCCCTGCCAGGT | Primer Extension |
| U1/17-39F | GGAGATACCATGATCACGAAGG | RT-qPCR |
| U1/58-80R | CATCCGGAGTGCAATGGATAAG | RT-qPCR |
| U2/8-19F | CTCGGCCTTTTGGCTAAGATCA | RT-qPCR |
| U2/62-84R | TCCTCGGATAGAGGACGTATCA | RT-qPCR |
Table 7: Sequence of oligonucleotides primers. List of primer names, sequences, and the technique the primers were used for.

Figure 1: Dup51 and Dup51p Minigene Reporters. (A) Schematic representation of the three-exon/two-intron minigene reporters, Dup51 and Dup51p. Locations of the Dup8f and Dup3r in exons 1 and 3, respectively, are indicated. (B) Predicted basepairing of the wildtype U1 snRNA and the U1-5a variant with the 5'-splice site sequences of Dup51 and Dup51p reporter transcripts. Please click here to view a larger version of this figure.
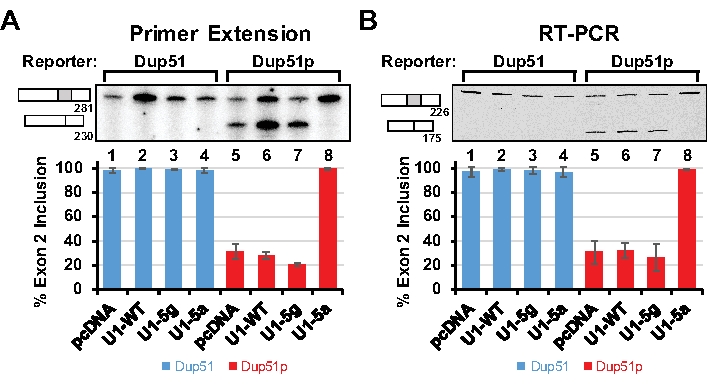
Figure 2: Analysis of Spliced Reporter Transcripts. HeLa cells were cotransfected with the Dup51 or Dup51p minigene plasmids and pcDNA control or pNS6U1 plasmids for U1-WT, U1-5g, or U1-5a snRNAs. (A) Primer extension analysis with 32P-labeled Dup3r primer demonstrating the splicing of the Dup51 and Dup51p minigene transcripts. The mRNA products are indicated to the left of the gel image. The percentage of the full-length product (± s.d., n = 3) was calculated from band intensities of the two mRNA isoforms and is represented in the graphs below. (B) RT-PCR analysis with Dup8f and Cy5-Dup3r primers on cDNA prepared from total RNA extracted from HeLa cells demonstrating splicing of the Dup51 or Dup51p minigene transcripts. The mRNA products are indicated to the left of the gel image. The percentage of the full-length product (± s.d., n = 3) was calculated from band intensities of the two mRNA isoforms and is represented in the graphs below. Please click here to view a larger version of this figure.
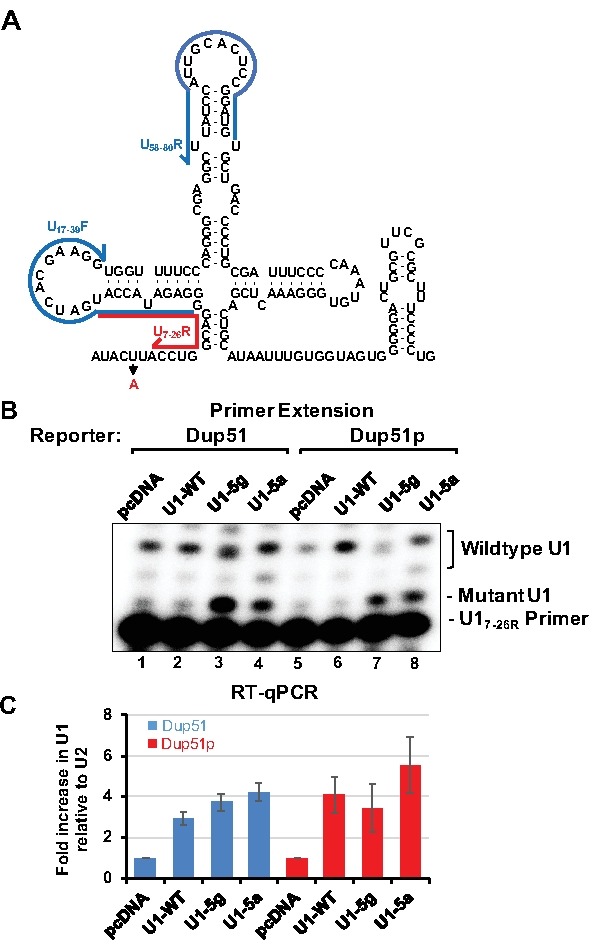
Figure 3: Analysis of the variant U1-5a snRNA. HeLa cells were cotransfected with the Dup51 or Dup51p minigene plasmids and pcDNA control or pNS6U1 plasmids for U1-WT, U1-5g, or U1-5a snRNAs. (A) A two-dimensional representation of the U1 snRNA sequence. Position of the U17-26R primer, used for primer extension analysis of U1 snRNA, is indicated by the red line. Position of U1/17-39F and U1/58-80R primers, used for RT-qPCR of U1 snRNA, is indicated by the blue lines.(B) Primer extension analysis with 32P-labeled U17-26 to detect the expression of the wildtype U1 snRNA, and the U1-5g and U1-5a variants. Positions of the products formed from U1-WT and the variants snRNAs are indicated. (C) RT-qPCR analysis of the levels of U1 snRNA expression in Hela cells. The fold change in U1-snRNA expression was calculated relative to the pcDNA negative control and normalized to U2-snRNA, fold change (± s.d.; n = 3) in U1 is graphed. Please click here to view a larger version of this figure.
