This tutorial provides an overview for biologists and algorithm design beginners on how to construct an easy-to-use deep learning framework for biological sequence classification in metagenomic data. This tutorial aims to provide intuitive understanding of deep learning and address the challenge that beginners often have difficulty installing the deep learning package and writing the code for the algorithm. For some simple classification tasks, users can use the framework to perform the classification tasks.
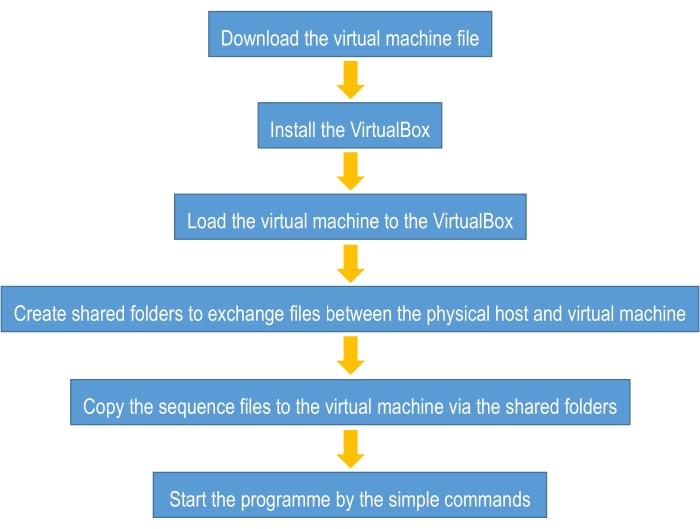
Considering that many biologists are not familiar with the command line of the Linux operating system, we preinstalled all the dependent software in a virtual machine. In this way, the user can directly run the code in the virtual machine following the protocol mentioned above. Additionally, if users are familiar with the Linux operating system and Python programming, they can also run this protocol directly on the server or local PC. In this way, the user should preinstall the following dependent software:
Python 2.7.12 (https://www.python.org/)
Python packages:
numpy 1.13.1 (http://www.numpy.org/)
h5py 2.6.0 (http://www.h5py.org/)
TensorFlow 1.4.1 (https://www.tensorflow.org/)
Keras 2.0.8 (https://keras.io/)
MATLAB Component Runtime (MCR) R2018a (https://www.mathworks.com/products/compiler/matlab-runtime.html)
The manual of our previous work3 has a brief description of the installation. Note that the version number of each package corresponds to the version that we used in the code. The advantage of running the code in the server or local PC without the virtual machine is that the code can speed up with a GPU in this way, which can save much time in the training process. In this way, the user should install the GPU version of TensorFlow (see the manual of previous work3).
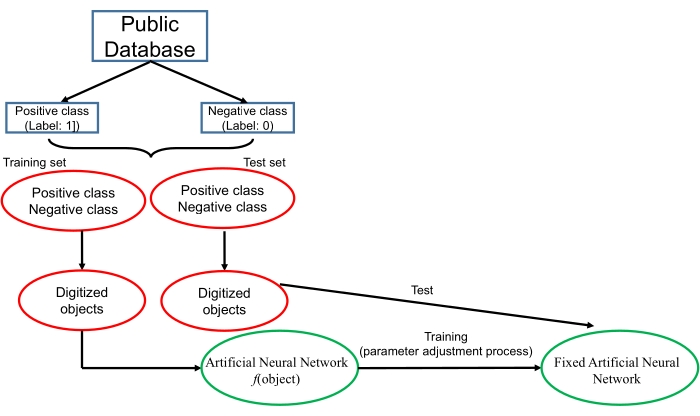
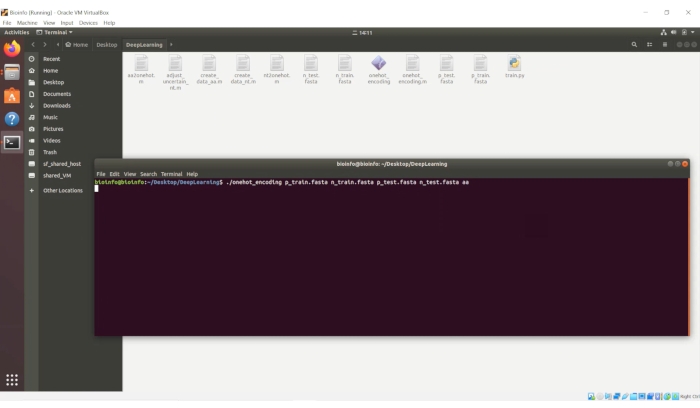
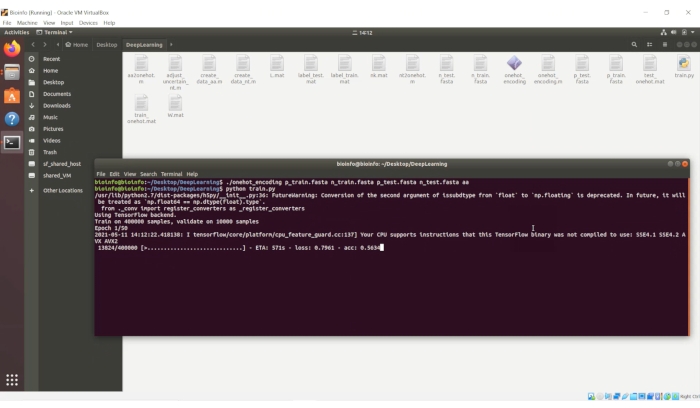
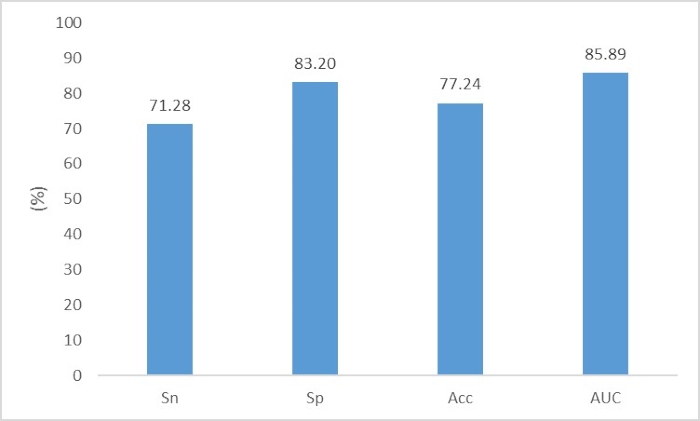
Some of the critical steps within the protocol are described as follows. In step 4.1, the file names of "p_train.fasta", "n_train.fasta", "p_test.fasta" and "n_test.fasta" should be replaced by the used file names. The order of these four files in this command cannot be changed. If the files contain amino acid sequences, the last parameter should be "aa"; if the files contain nucleic acid sequences, the last parameter should be "nt". This command uses the "one-hot" encoding form to digitize the biological sequences. An introduction of the "one-hot" encoding form is provided in the Supplementary Material. In step 5.1, because the virtual machine cannot be sped up with the GPU, this process may take a few hours or several days, depending on the data size. The progress bars for each iteration epoch are shown in the terminal. We set the number of epochs to 50, and thus, a total of 50 progress bars will be displayed when the training process is finished. When the test process is finished, the accuracy for the test set will be displayed in the terminal. In the "DeepLearning" folder of the virtual machine, a file named "predict.csv" will be created. This file contains all the prediction scores for the test data. The order of these scores corresponds to the sequence order in "p_test.fasta" and "n_test.fasta" (the first half of these scores corresponds to "p_test.fasta", while the second half of these scores corresponds to "n_test.fatsa"). If users want to make predictions for the sequences whose true classes are unknown, they can also deposit these unknown sequences either in the "p_test.fasta" or "n_test.fasta" file. In this way, the scores of these unknown sequences will also be displayed in the "predict.csv" file, but the "accuracy" display in the terminal does not make sense. This script employs a convolutional neural network to perform the classification. The structure of the neural network and the code for the neural network are shown in the Supplementary Material.
One of the characteristics of deep learning is that many parameter settings require some experience, which can be a major challenge for beginners. To avoid beginner apprehension caused by a large number of formulas, we do not focus on the mathematical principles of deep learning, and in the virtual machine, we do not provide a special parameter setting interface. Although this may be a good choice for beginners, inappropriate parameter selection may also lead to a decline in precision. To allow beginners to better experience how to modify the parameters, in the script "train.py", we add some comments to the related code, and users can modify the related parameters, such as the number of convolution kernels, to see how these parameters affect the performance.
Additionally, many deep learning programs should be run under a GPU. However, configuring the GPU also requires some computer skill that may be difficult for non-computer professionals; therefore, we choose to optimize the code in a virtual machine.
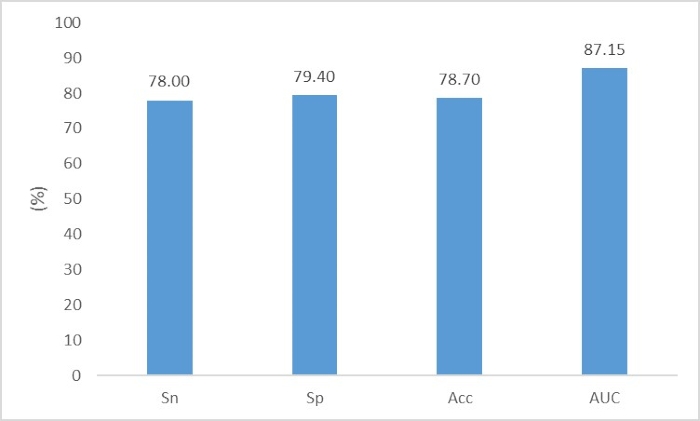
When solving other sequence classification tasks based on this guideline, users need only replace the four sequence files with their own data. For example, if users need to distinguish plasmid-derived and chromosome-derived sequences in metagenomic data, they can directly download plasmid genomes (https://ftp.ncbi.nlm.nih.gov/refseq/release/plasmid/) and bacterial chromosome genomes (https://ftp.ncbi.nlm.nih.gov/refseq/release/bacteria/) from the RefSeq database and separate the genomes into a training set and test set. It is worth noting that DNA sequences in metagenomic data are often fragmented rather than complete genomes. In such cases, users can use the MetaSim13 tool to extract the DNA fragment from the complete genome. MetaSim is a user-friendly tool with a GUI interface, and users can finish most operations using the mouse without typing any command on the keyboard. To simplify the operation for beginners, our tutorial is designed for a two-class classification task. However, we need to perform multiclassification in many tasks. In such cases, beginners can try to separate the multiclassification task into several two-class classification tasks. For example, to identify the phage host, Zhang et al. constructed 9 two-class classifiers to identify whether a given phage sequence can infect a certain host.
The homepage of this tutorial is deposited on the GitHub site https://github.com/zhenchengfang/DL-VM. Any update of the tutorial will be described on the website. Users can also raise their questions about this tutorial on the website.