Monitoring Leucine-Rich Repeat Containing 8 Channel (LRRC8/VRAC) Activity Using Sensitized-Emission F
Summary
Electrophysiology is the gold standard for investigating ion channel activity. However, there are plenty of alternative approaches, including optical methods. Here, we describe a method to monitor the activity of the leucine-rich repeat containing 8 channel (LRRC8)-formed anion channels using an inter-subunit Förster resonance energy transfer (FRET)-based method.
Abstract
Members of the LRRC8 protein family form heteromeric ion and osmolyte channels with roles in numerous physiological processes. As volume-regulated anion channels (VRACs)/volume-sensitive outwardly rectifying channels (VSORs), they are activated upon osmotic cell swelling and mediate the extrusion of chloride and organic osmolytes, leading to the efflux of water and hence cell shrinkage. Beyond their role in osmotic volume regulation, VRACs have been implicated in cellular processes such as differentiation, migration, and apoptosis. Through their effect on membrane potential and their transport of various signaling molecules, leucine-rich repeat containing 8 (LRRC8) channels play roles in neuron-glia communication, insulin secretion, and immune response. The activation mechanism has remained elusive. LRRC8 channels, like other ion channels, are typically studied using electrophysiological methods. Here, we describe a method to detect LRRC8 channel activation by measuring intra-complex sensitized-emission Förster resonance energy transfer (SE-FRET) between fluorescent proteins fused to the C-terminal leucine-rich repeat domains of LRRC8 subunits. This method offers the possibility to study channel activation in situ without exchange of the cytosolic environment and during processes such as cell differentiation and apoptosis.
Introduction
Ion channels comprised of heteromers of leucine-rich repeat containing 8 (LRRC8) family proteins are found throughout vertebrate cells, participating in a wide range of physiological functions1,2. These LRRC8 channels, first identified as volume-regulated anion channels (VRACs) or volume-sensitive outwardly rectifying channels (VSOR), play a crucial role in cellular regulatory volume decrease3,4. They facilitate the expulsion of chloride ions and organic osmolytes, which is followed by water efflux in response to osmotic swelling. Beyond their role in osmotic stress response, their role in cellular volume regulation has been linked to cell proliferation and migration, apoptosis, spermiogenesis, and epithelial integrity5,6,7. Alteration of the membrane potential upon LRRC8/VRAC activation has been shown to contribute to myotube differentiation8 and insulin secretion by pancreatic β-cells9,10,11. Furthermore, LRRC8 channels conduct a variety of organic osmolytes such as purinergic signaling molecules ATP and cGAMP or the excitatory amino acid glutamate, placing these channels in cell-cell communication in the immune system or glia-neuron interaction12,13,14,15,16,17,18,19,20,21,22. Even xenobiotics, such as the dye fluorescein, the antibiotic blasticidin S or the anticancer drug cisplatin, are conducted by LRRC8 channels23,24,25.
There are numerous reports on the signal transduction leading to LRRC8/VRAC activation26,27,28. However, the mechanism remains unclear, and the literature presents a broad range of potential mechanisms that could depend on the specific physiological process. These include changes in cytosolic ion strength, interaction with the cytoskeleton, membrane composition, G proteins, the redox state, and phosphorylation cascades2,27,29,30,31.
LRRC8/VRAC channels contain LRRC8A as an essential subunit3,4 that must heteromerize with at least one of its paralogues LRRC8B-E to form physiologically functional channels4,14,32. The subunit composition determines biophysical properties of the channel, such as rectification and depolarization-dependent inactivation4,29,32,33,34, substrate specificity15,17,20,21,24,35, and some activation pathways36,37. Cryo-electron microscopy (cryo-EM) structures show that LRRC8A homomers, as well as heteromers, assemble as hexamers38,39,40, while LRRC8A/LRRC8C chimeras that form functional channels are heptamers41. The N-terminal part of all LRRC8 proteins comprises four transmembrane helices, and the C-terminal part contains a domain with leucine-rich repeats (LRRD). The available LRRC8 complex structures provide evidence that the LRRDs, which stretch into the cytosol3,4,23, may undergo conformational rearrangements during channel gating34,42,43. This notion is corroborated by the finding that C-terminal fusion of fluorescent proteins results in basal channel activity14 and that binding of nanobodies to the domains can modulate channel activity44. Moreover, conformational alterations of the C-termini were shown by intra-complex Förster resonance energy transfer (FRET)45.
The most common method to study ion channel activity is electrophysiological measurements46, which were extensively applied in the investigation of VRACs before their molecular identification47. However, there are various additional ways to monitor VRAC activity indirectly, including the measurement of its conducted substrates -halide ions or organic osmolytes- or its effect on cell volume48. In fact, the identification of LRRC8 proteins as VRAC relied on an assay based on the quenching of a halide-sensitive fluorescent protein49 by iodide entering the cell through activated VRACs3,4. Another method to monitor LRRC8/VRAC channel activity makes use of the movement of the cytosolic domains which can be observed, as in other ion channels50,51,52,53, by changes in FRET45. To this end, fluorescent proteins that serve as FRET pairs, such as cyan-fluorescent protein (CFP)/mCerulean3 as donor and yellow-fluorescent protein (YFP)/mVenus as acceptor, were fused to the C-termini of the LRRC8 proteins (Figure 1). Intra-complex FRET between LRRC8 subunits was shown by acceptor photobleaching experiments45. Avoiding the destructive photobleaching method, FRET changes over time were monitored by sensitized-emission FRET (SE-FRET), where basically the sensitized emission of the acceptor upon excitation of the donor due to the overlap of the emission spectrum of the donor with the excitation spectrum of the acceptor is measured. Application of extracellular hypotonicity, a stimulus for LRRC8/VRAC activation, resulted in a reversible reduction in SE-FRET intensity45. Importantly, simultaneous whole-cell patch-clamp measurements and FRET monitoring during hypotonic treatment showed that this reduction in FRET indeed mirrored LRRC8/VRAC activation45. This method, which avoids disrupting the plasma membrane or altering the intracellular environment by pipette solution, offers an alternative for monitoring LRRC8/VRAC activity. It is particularly useful in physiological settings where maintaining the native cytosol is crucial, subcellular resolution is necessary, or prolonged observation of channel activity is required.
Here, we present a protocol to study LRRC8/VRAC with such a FRET-based read-out. The protocol depicts how to handle and transfect cells, acquire sample and control images, analyze the data, and calculate sensitized emission FRET (SE-FRET) values.
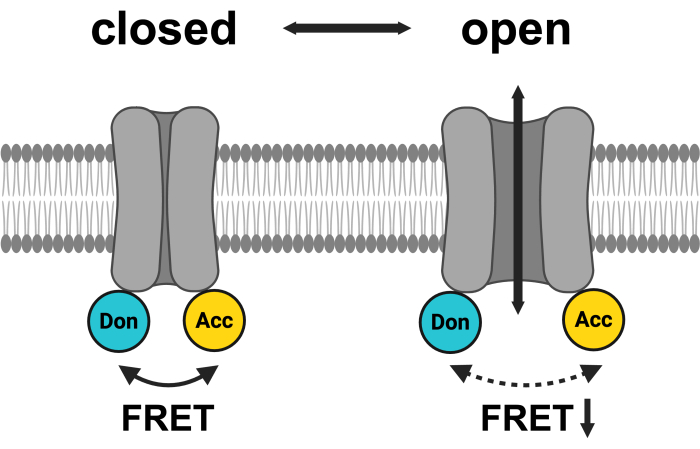
Figure 1: Schematic of the LRRC8 FRET pair system. mCerulean3 is shown in cyan, and mVenus is shown in yellow. Following VRAC opening, the distance (and/or the spatial orientation) between the fluorophores changes, resulting in a reduced energy transfer between donor (Don) and acceptor (Acc) and, in turn, lowering the observed FRET. Created with BioRender.com. Please click here to view a larger version of this figure.
Protocol
Representative Results
Discussion
FRET microscopy is a well-established, widely used technique to study the interaction between proteins. Hence, FRET-based methods can be applied in many laboratories of variable expertise. Conformational rearrangements during gating have been monitored for a broad range of ion channels using FRET-based assays (for examples, see references34,50,51,52,53,60,61,62,63,64,65,66,67), in some cases combined with electrophysiology in patch-clamp fluorometry68,69,70,71. FRET can be used to study the structure-function relationships of these ion channels or to monitor their activity independent of ion transport. The method presented here can have clear advantages over electrophysiology as it allows monitoring of the activity of LRRC8/VRAC channels in situ.
Critical steps in the protocol include plating the cells to reach optimal confluency for transfection and imaging, which ideally facilitates easy cell distinction for later analysis. Effective co-transfection of the different subunits is crucial for correct subcellular localization; for example, an excess of the non-LRRC8A subunit will lead to enhanced endoplasmic reticulum (ER) localization4. Therefore, plasmid ratios may need to be adjusted. Depending on the system, newly generated FRET pairs should be verified, e.g., by acceptor bleaching. Binning and exposure time must be balanced against each other to enable optimal temporal and spatial resolution for the research question. Binning enables shorter exposure times and hence reduces potential bleaching of the FRET sensor while decreasing spatial resolution. Therefore, if the experimental setup requires, e.g., subcellular discrimination of LRRC8/VRAC activity, binning should be avoided. The research question equally determines the number and interval of cycles in a time-lapse series. The interval is only relevant if the kinetics of the FRET changes (and hence LRRC8/VRAC activation/inactivation) are required; otherwise, simple "before-and-after" recordings can also be performed. The length of the experiment depends on the physiological process. Ideally, LRRC8/VRAC activity upon stimuli should be monitored until SE-FRET has stabilized. These factors can be determined in pilot experiments. Correction factors to calculate the real SE-FRET signal must be determined for all conditions. Incorrectly determined correction factors may lead to an over- or underestimation of the SE-FRET intensities. Lastly, after establishing a stable baseline, the time interval between images has to be short enough to capture the physiological process of interest.
The method bears some limitations. One of them is that changes in inter-LRRC8 FRET intensities while reflecting movements of the LRRDs, do not necessarily correspond to ion or osmolyte transport through the pore. This is clear from the FRET changes observed with LRRC8A homomers45 despite their minimal currents4,32,72. Pore blockers of LRRC8/VRAC channels may not affect the FRET signal, rendering this method unsuitable for the search for specific channel modulators. Moreover, the expression levels of the overexpressed LRRC8 proteins could affect the physiological processes that are observed, especially as the C-terminally tagged LRRC8 proteins display basal activity14.
An aspect that can be considered a limitation or an advantage depending on the particular research question is that in this method, only the ectopically expressed LRRC8 subunits are selectively measured. So, background levels of endogenous proteins hardly interfere with the measurements. On the other hand, the overexpressed proteins may not behave like the endogenous LRRC8 channels with potentially different subunit composition and stoichiometry. For example, various stimuli such as oxidation may have opposing modulatory effects on differently composed LRRC8 channels36. By altering the ratios between co-expressed subunits, their stoichiometry, and overall ion conductance can be adjusted14,73, but their native composition, with likely often more than two paralogs within one complex21, is not clear and may vary between cell types74,75,76. Furthermore, the fusion of fluorescent proteins to the cytosolic C-termini of LRRC8 proteins was shown to increase basal LRRC8/VRAC channel activity in Xenopus oocytes14, likely because the large tags modulate the conformation of the LRRDs, which may govern channel opening14,44,45. Therefore, the size of the fluorescent proteins, the linker, and their orientation may not only affect FRET efficiency but also channel activity. However, importantly, VRAC channels of LRRC8 proteins fused with fluorescent proteins remained responsive to hypotonic stimulation14, enabling their use as FRET sensors45.
Advantages of this non-invasive method to monitor LRRC8/VRAC channel activity by light microscopy compared to other methods comprise: (i) It allows observing LRRC8/VRAC within cells or compartments typically inaccessible for electrophysiology. This includes intracellular organelles on which LRRC8 complexes can be found or targeted to45,77,78. (ii) The cytosolic composition remains unaltered by the method, whereas during whole-cell patch-clamp measurements, the cytosol is largely replaced by pipette solution, which may affect signaling pathways as observed with phorbol-12-myristate-13-acetate (PMA)-induced LRRC8/VRAC activation45. (iii) It offers the possibility to observe LRRC8/VRAC activation with subcellular resolution, such as distinguishing activity at the leading and trailing edges during cell migration, where -restricted to confined spaces- VRAC has been implicated79,80. (iv) It enables continuous monitoring of LRRC8/VRAC activity during extended physiological processes such as myocyte differentiation56.
While there are limitations and challenges with this method, it holds promise for further exploration, including potential applications in animal models. In combination with other methods to study this ion and osmolyte channel family, this FRET-based assay may contribute significantly to unraveling activation mechanisms and exploring the diverse physiological functions of LRRC8 channels in their native environments.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
We thank C.F. Kaminski for the kind gift of the plasmid encoding the CFP-18aa-YFP construct, A. Klemmer for technical assistance, and all current and former members of the Stauber laboratory who contributed to the development of this method.
Materials
| 0.05% Trypsin-EDTA | gibco | 25300-054 | |
| Camera DFC9000GTC | Leica | 11547007 | |
| CFP-18aa-YFP | N/A | N/A | Elder et al. 2009 PMCID: PMC2706461; Gift from C.F. Kaminski (University of Cambridge, UK) |
| Cycloheximide (CHX) | Sigma-Aldrich | 66-81-9 | |
| D(-)-Mannitol | Carl Roth | 4175.1 | |
| D(+)-Glucose | Carl Roth | HN06.1 | |
| DMEM (Dulbeccos Modified Eagle Medium) | PAN-Biotech | P04-03590 | |
| DPBS (Dulbecco's Phosphate Buffer Saline) | PAN-Biotech | P04-36500 | |
| Emission filter wheel (460/80, 535/70, 590/50, 642/80, 100%) | Leica | 11525480 | |
| FBS (Fetal Bovine Serum) | PAN-Biotech | P30-3302 | |
| Filter cube CYR71010 | Leica | 11525416 | |
| FuGENE | Promega | E2691 | |
| Glas Bottom Culture Dishes 35 mm | MatTek | P35G-0-10-C | |
| HeLa cells | Leibniz Forschungsinstitut DSMZ | ACC 57 | Mammalian cervix carcinoma/ Obtained from Leibniz Forschungsinstitut DSMZ |
| HEPES | Carl Roth | 9105.4 | |
| ibidi µ-Disch 35 mm | ibidi | 81156 | |
| KCl (Potassium chloride) | Carl Roth | 6781.1 | |
| LAS X FRET Wizard | Leica | 11640862 | |
| Light source LED8 | Leica | 11504256 | |
| LRRC8A-mCerulean3 | N/A | N/A | König et al. 2019 |
| LRRC8E-mVenus | N/A | N/A | König et al. 2019 |
| Luna-II Automated Cell Counter | logos biosystems | L40002 | |
| Luna-II Cell Counter Slides | logos biosystems | L12001 | |
| MgCl2 (Magnesium chloride) | Carl Roth | KK36.1 | |
| Microscope THUNDER Imager live cell | Leica | 11525681 | |
| NaCl (Sodium chloride) | Carl Roth | 9263 | |
| Objective HC PL APO 63x/1.40 OIL | Leica | 11506349 | |
| Opti-Minimal Essential Medium (MEM) | gibco | 11058 | |
| Osmometer OM807 | Vogel | V04807 | |
| Penicillin Streptomycin (Pen Step) | gibco | 15070-063 | |
| Trypan blue solution (0,4%) | Sigma | T8154 | |
| Tumor necrosis factor (TNF)-a | Sigma-Aldrich | 94948-59-1 | |
| Valve Controlled Gravity Perfusion System | ALA Scientific Instruments | VC3-4xG |
Riferimenti
- Jentsch, T. J. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat Rev Mol Cell Biol. 17 (5), 293-307 (2016).
- Chen, L., et al. More than just a pressure relief valve: physiological roles of volume-regulated LRRC8 anion channels. Biol Chem. 400 (11), 1481-1496 (2019).
- Qiu, Z., et al. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell. 157 (2), 447-458 (2014).
- Voss, F. K., et al. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science. 344 (6184), 634-638 (2014).
- Hoffmann, E. K., Schettino, T., Marshall, W. S. The role of volume-sensitive ion transport systems in regulation of epithelial transport. Comp Biochem Physiol A Mol Integr Physiol. 148 (1), 29-43 (2007).
- López-Cayuqueo, K. I., et al. Renal deletion of LRRC8/VRAC channels induces proximal tubulopathy. J Am Soc Nephrol. 33 (8), 1528-1545 (2022).
- Lück, J. C., Puchkov, D., Ullrich, F., Jentsch, T. J. LRRC8/VRAC anion channels are required for late stages of spermatid development in mice. J Biol Chem. 293 (30), 11796-11808 (2018).
- Chen, L., Becker, T. M., Koch, U., Stauber, T. The LRRC8/VRAC anion channel facilitates myogenic differentiation of murine myoblasts by promoting membrane hyperpolarization. J Biol Chem. 294 (39), 14279-14288 (2019).
- Best, L., Brown, P. D., Sener, A., Malaisse, W. J. Electrical activity in pancreatic islet cells: The VRAC hypothesis. Islets. 2 (2), 59-64 (2010).
- Kang, C., et al. SWELL1 is a glucose sensor regulating beta-cell excitability and systemic glycaemia. Nat Commun. 9 (1), 367 (2018).
- Stuhlmann, T., Planells-Cases, R., Jentsch, T. J. LRRC8/VRAC anion channels enhance beta-cell glucose sensing and insulin secretion. Nat Commun. 9 (1), 1974 (2018).
- Hisadome, K., et al. Volume-regulated anion channels serve as an auto/paracrine nucleotide release pathway in aortic endothelial cells. J Gen Physiol. 119 (6), 511-520 (2002).
- Burow, P., Klapperstück, M., Markwardt, F. Activation of ATP secretion via volume-regulated anion channels by sphingosine-1-phosphate in RAW macrophages. Pflügers Arch. 467 (6), 1215-1226 (2015).
- Gaitán-Peñas, H., et al. Investigation of LRRC8-mediated volume-regulated anion currents in Xenopus oocytes. Biophys J. 111 (7), 1429-1443 (2016).
- Lahey, L. J., et al. LRRC8A:C/E heteromeric channels are ubiquitous transporters of cGAMP. Mol Cell. 80 (4), 578-591.e5 (2020).
- Chen, X., et al. Regulation of anion channel LRRC8 volume-regulated anion channels in transport of 2’3′-cyclic GMP-AMP and cisplatin under steady state and inflammation. J Immunol. 206 (9), 2061-2074 (2021).
- Zhou, C., et al. Transfer of cGAMP into bystander cells via LRRC8 volume-regulated anion channels augments STING-mediated interferon responses and anti-viral immunity. Immunity. 52 (5), 767-781.e6 (2020).
- Feustel, P. J., Jin, Y., Kimelberg, H. K. Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke. 35 (5), 1164-1168 (2004).
- Mongin, A. A. Volume-regulated anion channel–a frenemy within the brain. Pflügers Arch. 468 (3), 421-441 (2016).
- Schober, A. L., Wilson, C. S., Mongin, A. A. Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes. J Physiol. 595 (22), 6939-6951 (2017).
- Lutter, D., Ullrich, F., Lueck, J. C., Kempa, S., Jentsch, T. J. Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J Cell Sci. 130 (6), 1122-1133 (2017).
- Yang, J., et al. Glutamate-releasing SWELL1 channel in astrocytes modulates synaptic transmission and promotes brain damage in stroke. Neuron. 102 (4), 813-827.e6 (2019).
- Lee, C. C., Freinkman, E., Sabatini, D. M., Ploegh, H. L. The protein synthesis inhibitor blasticidin s enters mammalian cells via leucine-rich repeat-containing protein 8D. J Biol Chem. 289 (24), 17124-17131 (2014).
- Planells-Cases, R., et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anticancer drugs. EMBO J. 34 (24), 2993-3008 (2015).
- Model, M. A., Nia, F. H., Zook, E., Hollembeak, J. E., Stauber, T. Uptake of fluorescein upon osmotic cell swelling is dependent on the volume-regulated anion channel VRAC/LRRC8. Paracelsus Proc Exp Med. 1 (1), 3-14 (2022).
- Stauber, T. The volume-regulated anion channel is formed by LRRC8 heteromers – molecular identification and roles in membrane transport and physiology. Biol Chem. 396 (9-10), 975-990 (2015).
- Bertelli, S., et al. Mechanisms of activation of LRRC8 volume regulated anion channels. Cell Physiol Biochem. 55 (S1), 41-56 (2021).
- Liu, T., Li, Y., Wang, D., Stauber, T., Zhao, J. Trends in volume-regulated anion channel (VRAC) research: visualization and bibliometric analysis from 2014 to 2022. Front Pharmacol. 14, 1234885 (2023).
- Strange, K., Yamada, T., Denton, J. S. A 30-year journey from volume-regulated anion currents to molecular structure of the LRRC8 channel. J Gen Physiol. 151 (2), 100-117 (2019).
- Osei-Owusu, J., Yang, J., Vitery, M. D. C., Qiu, Z. Molecular biology and physiology of volume-regulated anion channel (VRAC). Curr Top Membr. 81, 177-203 (2018).
- Friard, J., Laurain, A., Rubera, I., Duranton, C. LRRC8/VRAC channels and the redox balance: A complex Relationship. Cell Physiol Biochem. 55 (S1), 106-118 (2021).
- Syeda, R., et al. LRRC8 proteins form volume-regulated anion channels that sense ionic strength. Cell. 164 (3), 499-511 (2016).
- Ullrich, F., Reincke, S. M., Voss, F. K., Stauber, T., Jentsch, T. J. Inactivation and anion selectivity of volume-regulated anion channels (VRACs) depend on C-terminal residues of the first extracellular loop. J Biol Chem. 291 (33), 17040-17048 (2016).
- König, B., Stauber, T. Biophysics and structure-function relationships of LRRC8-formed volume-regulated anion channels. Biophys J. 116 (7), 1185-1193 (2019).
- Concepcion, A. R., et al. The volume-regulated anion channel LRRC8C suppresses T cell function by regulating cyclic dinucleotide transport and STING-p53 signaling. Nat Immunol. 23 (2), 287-302 (2022).
- Gradogna, A., Gavazzo, P., Boccaccio, A., Pusch, M. Subunit-dependent oxidative stress sensitivity of LRRC8 volume-regulated anion channels. J Physiol. 595 (21), 6719-6733 (2017).
- Bertelli, S., Zuccolini, P., Gavazzo, P., Pusch, M. Molecular determinants underlying volume-regulated anion channel subunit-dependent oxidation sensitivity. J Physiol. 600 (17), 3965-3982 (2022).
- Deneka, D., Sawicka, M., Lam, A. K. M., Paulino, C., Dutzler, R. Structure of a volume-regulated anion channel of the LRRC8 family. Nature. 558 (7709), 254-259 (2018).
- Kasuya, G., et al. Cryo-EM structures of the human volume-regulated anion channel LRRC8. Nat Struct Mol Biol. 25 (9), 797-804 (2018).
- Kefauver, J. M., et al. Structure of the human volume regulated anion channel. Elife. 7, e38461 (2018).
- Takahashi, H., Yamada, T., Denton, J. S., Strange, K., Karakas, E. Cryo-EM structures of an LRRC8 chimera with native functional properties reveal heptameric assembly. Elife. 12, e82431 (2023).
- Sawicka, M., Dutzler, R. Regulators of cell volume: The structural and functional properties of anion channels of the LRRC8 family. Curr Opin Struct Biol. 74, 102382 (2022).
- Kasuya, G., Nureki, O. Recent advances in the structural biology of the volume-regulated anion channel LRRC8. Front Pharmacol. 13, 896532 (2022).
- Deneka, D., et al. Allosteric modulation of LRRC8 channels by targeting their cytoplasmic domains. Nat Commun. 12 (1), 5435 (2021).
- König, B., Hao, Y., Schwartz, S., Plested, A. J., Stauber, T. A FRET sensor of C-terminal movement reveals VRAC activation by plasma membrane DAG signaling rather than ionic strength. Elife. 8, e45421 (2019).
- Hille, B. . Ion Channels of Excitable Membranes. , (2001).
- Pedersen, S. F., Okada, Y., Nilius, B. Biophysics and physiology of the volume-regulated anion channel (VRAC)/volume-sensitive outwardly rectifying anion channel (VSOR). Pflügers Arch. 468 (3), 371-383 (2016).
- Kolobkova, Y., Pervaiz, S., Stauber, T. The expanding toolbox to study the LRRC8-formed volume-regulated anion channel VRAC. Curr Top Membr. 88, 119-163 (2021).
- Galietta, L. J., Haggie, P. M., Verkman, A. S. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 499 (3), 220-224 (2001).
- Bykova, E. A., Zhang, X. D., Chen, T. Y., Zheng, J. Large movement in the C terminus of CLC-0 chloride channel during slow gating. Nat Struct Mol Biol. 13 (12), 1115-1119 (2006).
- Zheng, J., Zagotta, W. N. Gating rearrangements in cyclic nucleotide-gated channels revealed by patch-clamp fluorometry. Neuron. 28 (2), 369-374 (2000).
- Miranda, P., et al. State-dependent FRET reports calcium- and voltage-dependent gating-ring motions in BK channels. Proc Natl Acad Sci U S A. 110 (13), 5217-5222 (2013).
- Zachariassen, L. G., et al. Structural rearrangement of the intracellular domains during AMPA receptor activation. Proc Natl Acad Sci U S A. 113 (27), E3950-E3959 (2016).
- Markwardt, M. L., et al. An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. PLoS One. 6 (3), e17896 (2011).
- Nagai, T., et al. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 20 (1), 87-90 (2002).
- Chen, L., König, B., Stauber, T. LRRC8 channel activation and reduction in cytosolic chloride concentration during early differentiation of C2C12 myoblasts. Biochem Biophys Res Commun. 532, 482-488 (2020).
- Maeno, E., Ishizaki, Y., Kanaseki, T., Hazama, A., Okada, Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc Natl Acad Sci U S A. 97 (17), 9487-9492 (2000).
- Shimizu, T., Numata, T., Okada, Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl- channel. Proc Natl Acad Sci U S A. 101 (17), 6770-6773 (2004).
- Elder, A. D., et al. A quantitative protocol for dynamic measurements of protein interactions by Förster resonance energy transfer-sensitized fluorescence emission. Journal of the Royal Society Interface. 6 (Suppl 1), S59-S81 (2009).
- Glauner, K. S., Mannuzzu, L. M., Gandhi, C. S., Isacoff, E. Y. Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature. 402 (6763), 813-817 (1999).
- Dai, G., Aman, T. K., DiMaio, F., Zagotta, W. N. The HCN channel voltage sensor undergoes a large downward motion during hyperpolarization. Nat Struct Mol Biol. 26 (8), 686-694 (2019).
- Renart, M. L., et al. Conformational plasticity in the KcsA potassium channel pore helix revealed by homo-FRET studies. Sci Rep. 9 (1), 6215 (2019).
- Wang, S., et al. Potassium channel selectivity filter dynamics revealed by single-molecule FRET. Nat Chem Biol. 15 (4), 377-383 (2019).
- Harley, C. A., et al. Conformation-sensitive antibody reveals an altered cytosolic PAS/CNBh assembly during hERG channel gating. Proc Natl Acad Sci U S A. 118 (44), e2108796118 (2021).
- Han, S., et al. Structural dynamics determine voltage and pH gating in human voltage-gated proton channel. Elife. 11, e73093 (2022).
- Cullinan, M. M., Klipp, R. C., Camenisch, A., Bankston, J. R. Dynamic landscape of the intracellular termini of acid-sensing ion channel 1a. Elife. 12, e90755 (2023).
- Kim, J., Won, J., Chung, D. K., Lee, H. H. FRET analysis of the temperature-induced structural changes in human TRPV3. Sci Rep. 13 (1), 10108 (2023).
- Zheng, J., Zagotta, W. N. Patch-clamp fluorometry recording of conformational rearrangements of ion channels. Sci STKE. 2003 (176), PL7 (2003).
- Kusch, J., Zifarelli, G. Patch-clamp fluorometry: electrophysiology meets fluorescence. Biophys J. 106 (6), 1250-1257 (2014).
- Cowgill, J., Chanda, B. The contribution of voltage clamp fluorometry to the understanding of channel and transporter mechanisms. J Gen Physiol. 151 (10), 1163-1172 (2019).
- Bhat, S., Blunck, R. Characterising ion channel structure and dynamics using fluorescence spectroscopy techniques. Biochem Soc Trans. 50 (5), 1427-1445 (2022).
- Yamada, T., Figueroa, E. E., Denton, J. S., Strange, K. LRRC8A homohexameric channels poorly recapitulate VRAC regulation and pharmacology. Am J Physiol Cell Physiol. 320 (3), C293-C303 (2021).
- Yamada, T., Wondergem, R., Morrison, R., Yin, V. P., Strange, K. Leucine-rich repeat containing protein LRRC8A is essential for swelling-activated Cl- currents and embryonic development in zebrafish. Physiol Rep. 4 (19), e12940 (2016).
- Pervaiz, S., Kopp, A., von Kleist, L., Stauber, T. Absolute protein amounts and relative abundance of volume-regulated anion channel (VRAC) LRRC8 subunits in cells and tissues revealed by quantitative immunoblotting. Int J Mol Sci. 20 (23), 5879 (2019).
- Kern, D. M., et al. Structural basis for assembly and lipid-mediated gating of LRRC8A:C volume-regulated anion channels. Nat Struct Mol Biol. 30 (6), 841-852 (2023).
- Rutz, S., Deneka, D., Dittmann, A., Sawicka, M., Dutzler, R. Structure of a volume-regulated heteromeric LRRC8A/C channel. Nat Struct Mol Biol. 30 (1), 52-61 (2023).
- Li, P., et al. LRRC8 family proteins within lysosomes regulate cellular osmoregulation and enhance cell survival to multiple physiological stresses. Proc Natl Acad Sci U S A. 117 (46), 29155-29165 (2020).
- Kashyap, P., et al. An optogenetic method for the controlled release of single molecules. Nat Methods. 21 (4), 666-672 (2024).
- Liu, T., Stauber, T. The volume-regulated anion channel LRRC8/VRAC is dispensable for cell proliferation and migration. Int J Mol Sci. 20 (11), E2663 (2019).
- Zhang, Y., et al. Polarized NHE1 and SWELL1 regulate migration direction, efficiency and metastasis. Nat Commun. 13 (1), 6128 (2022).

.