Nonradioactive Assay to Measure Polynucleotide Phosphorylation of Small Nucleotide Substrates
Summary
This protocol describes a nonradioactive assay to measure kinase activity of polynucleotide kinases (PNKs) on small DNA and RNA substrates.
Abstract
Polynucleotide kinases (PNKs) are enzymes that catalyze the phosphorylation of the 5' hydroxyl end of DNA and RNA oligonucleotides. The activity of PNKs can be quantified using direct or indirect approaches. Presented here is a direct, in vitro approach to measure PNK activity that relies on a fluorescently-labeled oligonucleotide substrate and polyacrylamide gel electrophoresis. This approach provides resolution of the phosphorylated products while avoiding the use of radiolabeled substrates. The protocol details how to set up the phosphorylation reaction, prepare and run large polyacrylamide gels, and quantify the reaction products. The most technically challenging part of this assay is pouring and running the large polyacrylamide gels; thus, important details to overcome common difficulties are provided. This protocol was optimized for Grc3, a PNK that assembles into an obligate pre-ribosomal RNA processing complex with its binding partner, the Las1 nuclease. However, this protocol can be adapted to measure the activity of other PNK enzymes. Moreover, this assay can also be modified to determine the effects of different components of the reaction, such as the nucleoside triphosphate, metal ions, and oligonucleotides.
Introduction
Polynucleotide kinases (PNK) play critical roles in many DNA and RNA processing pathways, such as DNA repair and ribosome assembly1,2,3,4,5. These fundamental enzymes catalyze the transfer of the terminal (gamma) monophosphate from a nucleoside triphosphate (NTP, most often ATP) to the 5' hydroxyl end of a nucleotide substrate. One of the most well characterized PNKs is bacteriophage T4 PNK, which has broad substrate specificity and is heavily utilized by molecular biology labs for incorporating radioactive isotope labels onto the 5 terminus of a DNA or RNA substrate6,7,8,9,10,11,12. Another example of a PNK enzyme is CLP1, which is found in Eukarya, Eubacteria, and Archaea, and is implicated in several RNA processing pathways4,13,14,15.
Historically, most assays that measure polynucleotide kinase activity are dependent upon radioactive isotope labeling and subsequent autoradiography5,16. In recent years a number of additional assays have been developed to measure PNK activity, including single molecule approaches, microchip electrophoresis, molecular beacons, as well as colorimetric and luminescence-based assays17,18,19,20,21,22. While many of these new approaches provide enhanced detection limits and avoid the use of radioactivity, each has drawbacks, such as cost, reliance on immobilized resin, and limitations in substrate choice.
Grc3 is a polynucleotide kinase that plays a pivotal role in the processing of pre-ribosomal RNA2,3,23. Grc3 forms an obligate complex with the endoribonuclease Las1, which cleaves the Internal Transcribed Spacer 2 (ITS2) of the pre-ribosomal RNA3. Cleavage of the ITS2 by Las1 generates a product harboring a 5' hydroxyl that is subsequently phosphorylated by the Grc3 kinase3. To investigate the nucleotide and substrate specificity of Grc3, an inexpensive assay that allowed testing of different oligonucleotide substrates was required. Therefore, a PNK phosphorylation assay using fluorescently-labeled substrates was developed. This assay was successfully used to determine that Grc3 can utilize any NTP for phosphoryl transfer activity, but favors ATP24. This protocol adapts the original assay to measure PNK activity of Grc3 on an RNA mimic of its pre-ribosomal RNA substrate (SC-ITS2, Table 1). One challenging aspect of this fluorescence-based approach is the reliance on large polyacrylamide gels to effectively resolve phosphorylated and nonphosphorylated substrates. The protocol provides specific details on how to pour these large gels and avoid common pitfalls when doing so.
Working with RNA requires particular care because it is strongly susceptible to degradation. There are simple preventative steps one can take to limit ribonuclease contamination. A separate RNA workstation that can be easily treated with an RNase inhibitor-containing cleaning agent is often helpful. Always wearing gloves when handling samples and use of RNase-free certified consumables is necessary. Because water is another common source of contamination, it is best to use freshly purified water and sterilize all solutions using a 0.22 μm filter.
Protocol
1. Preparation
- Prepare buffer and reagents.
- Make 1x Reaction Buffer by combining 20 µL of 1 M Tris (pH = 8.0), 40 µL of 5 M sodium chloride, 2.5 µL of 2 M magnesium chloride, 100 µL of 50% (v/v) glycerol, and RNase-free water to reach a total volume of 1 mL.
- Make urea loading dye by combining 4.8 g of urea, 200 µL of 1 M Tris (pH = 8.0), 20 µL of 0.5 M EDTA (pH = 8.0), 0.5 mL of 1% (w/v) bromophenol blue, and RNase-free water to reach a total volume of 10 mL.
- Make 10x TBE buffer by combining 108 g of Tris base, 55 g of boric acid, 40 mL of 0.5 M EDTA (pH = 8.0), and RNase-free water to reach a total volume of 1 L.
- Make 10% (w/v) ammonium persulfate (APS). Weigh 0.5 g of APS and add 3 mL of RNase-free water to dissolve the solid. Add RNase-free water to reach a total volume of 5 mL. Wrap the tube in foil and store the solution at 4 °C.
NOTE: Dissolved APS decays over time. Replace the 10% APS stock every 2 weeks.
- Obtain polynucleotide kinase enzyme.
- Acquire pure Las1-Grc3 PNK enzyme2 stored in Reaction Buffer (see step 1.1.1) in a stock concentration of 20 μM. The storage buffer will vary depending on the specific PNK.
- Prepare nucleic acid substrate.
NOTE: This protocol monitors 5'-phosphorylation of a 27 nt RNA substrate that harbors the Las1-Grc3 processing site (C2 site) contained within the Saccharomyces cerevisiae pre-ribosomal internal transcribed spacer 2 (SC-ITS2; see Table 1)2,25,26.- Chemically synthesize an RNA oligonucleotide substrate containing a 5'-hydroxyl end along with a 3'-fluorescent label. The fluorophore will be used for the visualization of the RNA. Ensure that the fluorophore is positioned on the RNA end that is not targeted for phosphorylation. As an alternative to commercial sources, internal and terminal fluorescent labeling of RNA substrates can be performed in-house using cost-effective protocols27.
- Remove excess fluorophore from the RNA labeling reaction by HPLC-purification28.
- Resuspend the lyophilized RNA using RNase-free water to 500 nM.
- Store RNA aliquots at -80 °C for long-term storage or -20 °C for short-term storage.
- Prepare the ATP concentration series.
- Make a 20 mM working stock of ATP using Reaction Buffer (see step 1.1.1). Use this working stock to generate four serial dilutions ranging from 20 mM-0.02 mM.
- To make the first dilution of the concentration series, mix 1 μL of the 20 mM ATP working stock with 9 μL of Reaction Buffer. This will result in a 10-fold dilution of the ATP with a final concentration of 2 mM.
- Use the 2 mM ATP stock generated in step 1.4.2 to make the next dilution in the concentration series. Mix 1 μL of the 2 mM ATP stock with 9 μL of Reaction Buffer. This will make a new ATP stock concentration of 0.2 mM.
- Continue diluting 1 μL of the previous ATP stock with 9 μL of Reaction Buffer to make the subsequent ATP stock in the concentration series.
2. In vitro RNA kinase reaction
NOTE: This assay can be used to measure several variables such as time, nucleotide levels, or enzyme concentration. The goal of this experiment is to assess the amount of phosphorylated RNA in the presence of constant Las1-Grc3 complex and varying ATP levels.
- For each RNA-enzyme kinase reaction, combine 1 μL of 500 nM RNA substrate, 8.3 μL of 130 nM Las1-Grc3, and 0.2 μL of 5 mM EDTA.
NOTE: If carrying out several reactions, prepare a master stock of the RNA-enzyme mixture and aliquot 9.5 μL of this master mix to each reaction tube. - Start the assay.
- Set the heat block to 37 °C.
- At 10 s intervals, mix 0.5 μL of one ATP substock from the ATP concentration series prepared in step 1.4 with one RNA-enzyme mixture and place the reaction in the 37 °C heat block.
NOTE: Add 0.5 μL of Reaction Buffer instead of ATP to one RNA-enzyme mixture for a PNK negative control. Use another necessary control as well (i.e., RNA substrate in the absence of PNK enzyme). - Incubate the reactions for 60 min at 37 °C.
NOTE: Fluorescently-labeled RNA is light sensitive; therefore, cover the reactions with foil. - Using the same order as in step 2.2.2, quench each reaction every 10 s by spiking the reaction with 10 μL of urea loading dye (see step 1.1.2).
NOTE: In addition to 4 M urea, proteinase K may be added at this step to degrade the enzyme. Follow the instructions provided by the supplier to determine the required protease amount and reaction conditions. - Use the quenched in vitro RNA kinase reactions immediately for downstream analysis (see section 3) or store at -20 °C to be analyzed at a later date.
3. Gel electrophoresis
- Prepare 15% acrylamide/8 M urea gel solution.
CAUTION: Acrylamide must be handled with care, because it is a neurotoxin. Always wear gloves, a lab coat, and goggles when handling it.- In a 150 mL glass beaker, combine 22.5 mL of premixed 40% acrylamide/bis-acrylamide 29:1 solution, 6 mL of 10x TBE (see step 1.1.3), 28.8 g of urea, and RNase-free water to a total volume of 59 mL. Gently stir the solution.
NOTE: Depending on the RNA substrate length, altering the percentage of polyacrylamide may improve the resolution between unphosphorylated and phosphorylated RNA. - To dissolve the urea, heat the solution in the microwave for 20 s, stir the liquid, and immediately place the solution back into the microwave for another 20 s. Gently stir the solution until the urea completely dissolves.
- Slowly cool the solution by placing the glass beaker into a shallow water bath containing cold water. Make sure that the level of cold water surrounding the glass beaker is above the level of solution inside the glass beaker. This will promote efficient heat transfer. Wait 5 min.
NOTE: Do not continue with this protocol if the glass beaker feels warm; the water should be below 25 °C. If it still feels warm after 5 min, then replace the water in the water bath with fresh cold water and wait another 5 min. - Filter and degas the solution using a 0.22 μm disposable filtration unit to remove particulates and microscopic air bubbles.
- In a 150 mL glass beaker, combine 22.5 mL of premixed 40% acrylamide/bis-acrylamide 29:1 solution, 6 mL of 10x TBE (see step 1.1.3), 28.8 g of urea, and RNase-free water to a total volume of 59 mL. Gently stir the solution.
- Pour the denaturing gel.
- Use soap and warm water to clean a short and long glass plate designed for gels with overall dimensions of 31.0 cm x 38.5 cm. Spray each glass plate with 95% ethanol and wipe the glass to remove any moisture.
NOTE: One of the two plates can be siliconized to prevent damage to the gel when separating the glass plate sandwich. However, this step is not necessary because this protocol is designed to visualize the RNA without separating the glass plates. - Place the long glass plate horizontally on top of a box so it is elevated off the benchtop.
CAUTION: Acrylamide is toxic, therefore the gel pouring station must be covered in bench paper that can absorb any spilled liquid and be immediately placed in a waste bag after the procedure is complete. - Place a clean 0.4 mm spacer along the long edges of the long glass plate.
- Lay the short glass plate atop the long plate and ensure the edges of the short plate, long plate, and spacers are aligned. Clamp each side using three evenly spaced metal clamps.
- Add 24 μL of TEMED to the 15% acrylamide/8 M urea solution prepared in step 3.1 and mix the solution.
- Add 600 μL of 10% (w/v) APS (see step 1.1.4) to the solution in step 3.2.5 and gently mix the solution.
- Immediately pour the solution between the glass plates.
NOTE: To avoid bubbles, tap the glass as the solution is poured. - Carefully add a clean, 0.4 mm, 32 well comb to the top of the glass plate sandwich.
- Allow a minimum of 30 min for the acrylamide to polymerize.
- Use soap and warm water to clean a short and long glass plate designed for gels with overall dimensions of 31.0 cm x 38.5 cm. Spray each glass plate with 95% ethanol and wipe the glass to remove any moisture.
- Run the denaturing gel.
- Set the heat block to 75 °C.
- Remove the metal clamps holding the glass plate sandwich together and thoroughly wash and dry the glass plate sandwich.
- Place the glass plate sandwich into the gel apparatus with the short plate facing inward.
- Prepare 0.5x TBE running buffer by combining 100 mL of 10x TBE with 1.9 L RNase-free water. Add 600 mL of the running buffer to the upper and lower chambers of the gel apparatus.
- Gently remove the comb and rinse the wells thoroughly using a syringe.
NOTE: This step is critical for removing urea from the wells. - Pre-run the gel for 30 min at 50 W. Voltage is approximately 2,000 V.
CAUTION: This gel apparatus operates at a high wattage and users should exhibit precaution. - Rinse the wells thoroughly using a syringe.
NOTE: This step is critical for even loading of sample into the wells. - Pulse spin the quenched reactions from step 2.2.5. Then incubate the tubes at 75 °C for 3 min. Repeat the pulse spin.
- Immediately load 10 μL of sample per well and run the gel for 3 h at 50 W.
NOTE: Fluorescently-labeled RNA is light sensitive; therefore, cover the gel apparatus with foil.
- Image the denaturing gel.
- Turn off the power supply and drain the upper chamber of the gel apparatus.
- Wash and dry the outer side of the glass plate sandwich using soap and water. Cover the glass plate sandwich with foil while transporting the gel.
- Mount the glass plate sandwich onto the stage of a laser scanner capable of quantitative and sensitive fluorescence detection.
NOTE: This gel is extremely thin for maximum resolution. For this reason, this protocol is designed to avoid the difficulties of removing the gel from the glass plate sandwich by directly visualizing the fluorescently-labeled RNA through the glass plates. The use of commercially available low-fluorescence glass plates will increase the captured signal by improving the signal-to-noise ratio. - Set the excitation and emission wavelengths of the laser scanner for the desired fluorophore.
NOTE: The optimal excitation and emission wavelengths may differ for specific fluorophores. For the FAM fluorophore, the excitation and emission wavelengths are 495 nm and 535 nm, respectively. - Define the area to be visualized on the laser scanner stage and image the gel according to the manufacturer's instructions (Figure 1).
4. Image analysis and signal quantification
- Load the digital gel image acquired in step 3.4.5 into image procesing software (see Table of Materials).
- Using the left button of the mouse, click and drag a rectangle to define a template box to mark the boundaries on the digital gel image that will be used to quantify each RNA band. The RNA band seen in the the reaction mixture set in the absence of PNK enzyme is usually a good band to generate this template.
NOTE: To avoid bias caused by background signal, it is critical that the area surrounding each RNA band being measured is identical. For this reason, it is best to use the same template box to demarcate each RNA band. - Open the ROI Manager under "Tools" and mark the position of the template box by clicking "Add".
NOTE: Quantitation programs can also draw boxes automatically following the software user manual. - Using the left button of the mouse, click and drag the template box over to the next RNA band and repeat step 4.3. Continute this process until the position of all the unphosphorylated and phosphorylated RNA bands in each reaction have been marked.
- Measure the integrated density within each box by clicking "Measure" in the ROI Manager.
- To calculate the relative amount of phosphorylated RNA in a reaction, divide the integrated density of the phosphorylated RNA band by the sum of the integrated densities of the unphosphorylated and phosphorylated RNA bands from the same reaction. This approach can also be applied to calculate the relative amount of unphosphorylated RNA.
- To visualize the accumulation of phosphorylated RNA product and the corresponding depletion of unphosphorylated RNA substrate across the ATP concentration series, plot the relative amount of RNA calculated in step 4.6 against the concentration of ATP.
Representative Results
A successful representative denaturing gel of a titration of ATP with a fixed amount of Las1-Grc3 complex is shown in Figure 1. Addition of enzyme resulted in Las1-mediated RNA cleavage of the SC-ITS2 RNA substrate, leading to a defined RNA fragment (5-OH C2 RNA). Upon the addition of ATP, the C2 RNA fragment was phosphorylated by Grc3 PNK (5-P C2 RNA). In denaturing gels the phosphorylated RNA migrates faster than its unphosphorylated counterpart. As shown in Figure 2, phosphorylation of the C2 RNA fragment could be visualized by plotting the relative amount of unphosphorylated and phosphorylated C2 RNA against the ATP concentration. Grc3 PNK also phosphorylated the 5-end of the uncut SC-ITS2 substrate, albeit at a lower rate. This confirms previous work suggesting Grc3 PNK shows substrate preference towards its C2 RNA substrate24. An unsuccessful denaturing gel is shown in Figure 3. This gel was unsuccessful because the 21 nt RNA substrate contained degradation products (Figure 3, first lane). These degradation products overlapped with the phosphorylated product and made it impossible to accurately quantify phosphorylation. In contrast, the shortest RNA degradation product (Figure 3, gray arrows) could be successfully analyzed because this area of the gel did not contain any additional RNA species that hindered accurate quantification of its phosphorylated counterpart. To avoid RNA degradation bands, keep the workspace and solutions RNase-free, and limit the number of RNA freeze-thaw cycles.
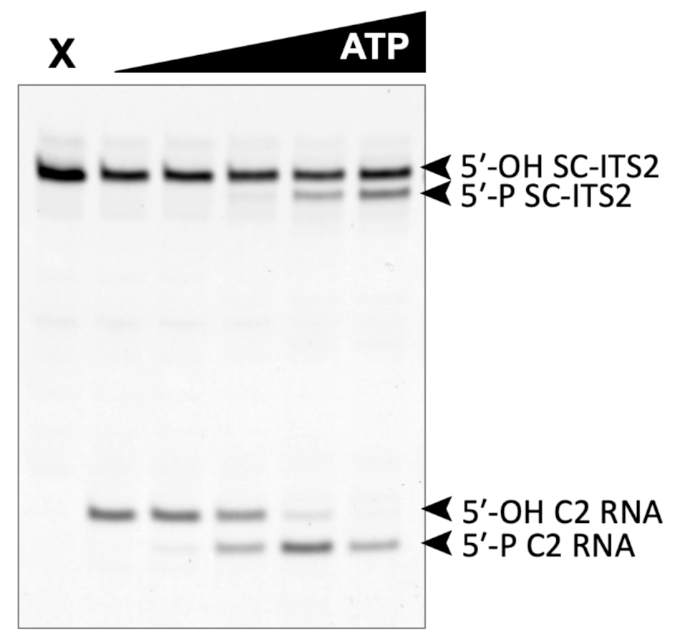
Figure 1: Example of denaturing gel analysis of phosphorylated RNA. In vitro RNA kinase assay of Las1-Grc3 (110 nM) incubated with 500 nM SC-ITS2 RNA. X marks the control reaction without Las1-Grc3 and the black triangle represents the titration of ATP from 0-10 mM. C2 RNA is the result of SC-ITS2 RNA cleavage by the Las1 nuclease and is the endogenous substrate for Grc3 PNK activity. Please click here to view a larger version of this figure.
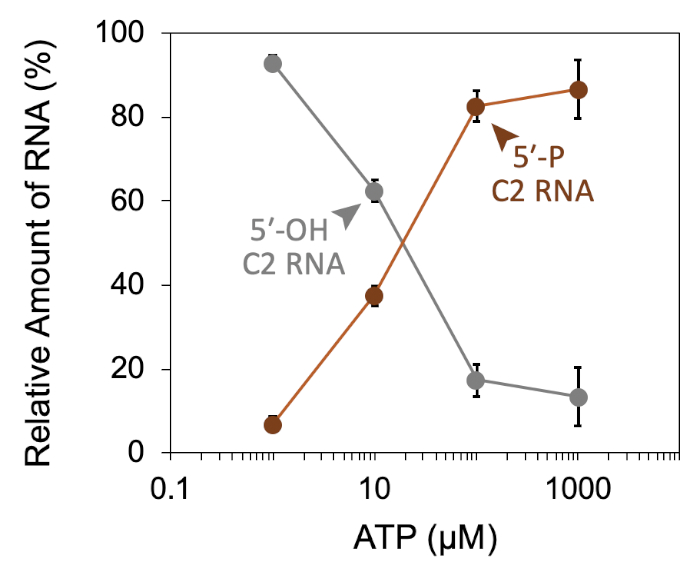
Figure 2: Quantification of RNA phosphorylation. Densiometric plot of Las1-Grc3 RNA kinase activity expressed as a percentage of unphosphorylated C2 RNA (grey line; 5-OH C2 RNA) and phosphorylated C2 RNA (brown line; 5-P C2 RNA) across an ATP concentration series. Error bars mark the standard deviation from three independent technical replicates. Please click here to view a larger version of this figure.
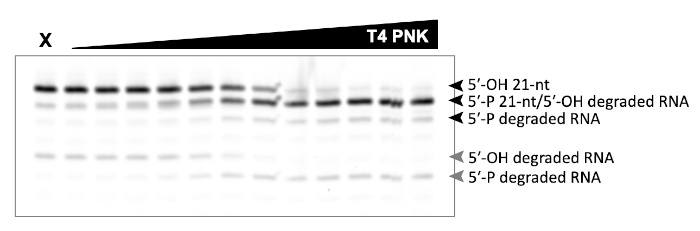
Figure 3: Example of an unsuccessful denaturing gel analysis of phosphorylated RNA. In vitro RNA kinase assay of T4 PNK (0-0.625 U) incubated with 5 μm 21 nt RNA. X marks the control reaction without T4 PNK. This RNA only control contains degraded RNA products that make it impossible to distinguish the 21 nt phosphorylated product from the degradation product (black arrows). Phosphorylation of the shortest RNA degradation product (gray arrows) can be analyzed, because there are no dedradation products that overlap with its phosphorylated counterpart. Please click here to view a larger version of this figure.
| Sequence (5'→3') | Oligo Code | Source | |
| SC-ITS2 | GUCGUUUUAGGUUUUAC CAACUGCGGC/36-FAM/ |
mp 911 | (Pillon et al. NSMB 2019)26 |
| C2 RNA | GGUUUUACCAACUG CGGC/36-FAM/ |
N/A | Las1 cleavage product of SC-ITS2 |
| 21-nt | ACGUACGCGGAA UACUUCGAA/36-TAMSp/ |
mp 596 | (Pillon et al. RNA, 2018)24 |
Table 1: Fluorescently-labeled RNA substrates.
Discussion
Described is an assay to measure kinase activity of Grc3 PNK on fluorescently-labeled nucleotide substrates. This protocol can be applied to characterize other PNK enzymes by adapting the Reaction Buffer and oligonucleotide substrate. For instance, the protocol calls for a trace amount of EDTA. The addition of EDTA is beneficial for two reasons: First, this approach favors magnesium-bound Grc3 by preventing the enzyme from binding to trace amounts of contaminating metals in the mixture. Second, a small amount of EDTA inhibits the activity of contaminating metal-dependent ribonucleases without disrupting the activity of the associated Las1 metal-independent ribonuclease. The concentration of EDTA may be altered depending on the source of the specific PNK. This assay also provides an advantage over traditional PNK phosphorylation assays that rely on oligonucleotides labeled with short half-life radioisotopes (i.e., 2 week half-life for phosphorus-32). This protocol can be adapted to measure specific enzyme activity and Michaelis-Menten kinetics as well as determine the dependence of phosphorylation on various parameters, such as the nature of the nucleotides, substrates, and metal ions.
This protocol relies on running large denaturing polyacrylamide gels in order to achieve sufficient resolution to distinguish an unphosphorylated substrate from its phosphorylated product. Pouring and handling these gels are critical steps in the protocol. For instance, the acrylamide solution must be heated to dissolve the urea and then cooled slowly before pouring the gel. Cooling this solution too quickly can lead to the formation of crystals. Care must also be taken to avoid introducing air bubbles and dust while pouring and setting the gel. Imaging the gel without removing the glass plates is recommended to avoid ripping the gel, which is thin and difficult to handle once removed from the glass plates.
This protocol can be adapted to measure the phosphorylation of different sized oligonucleotide substrates. This particular assay used a 15% acrylamide gel to achieve resolution of phosphorylation of 27 nt (SC-ITS2 RNA) and 18 nt (C2 RNA) substrates. The addition of one phosphate group to the 5-end of SC-ITS2 RNA produces a modest shift compared to the mobility change induced upon 5-phosphorylation of the shorter C2 RNA (Figure 1). This highlights a limitation in RNA length when using this technique. With longer RNA substrates, the contribution of a phosphate group to the overall molecular weight of the RNA species is diminished. For this reason, the percentage of acrylamide as well as the gel running time can be optimized to achieve resolution of different sized substrates29. In general, higher percentages of acrylamide are used for smaller oligonucleotide substrates while lower percentages of acrylamide (down to 8%) provide better resolution of larger oligonucleotide substrates.
The major limitation of this assay is that it is low-throughput. Pouring and running denaturing gels takes a significant amount of time, limiting the number of gels and samples that can be analyzed in a day. A future application to overcome this limitation could be the development of microfluidic chips able to resolve phosphorylated oligonucleotide products. Microfluidic-based gel electrophoresis has many advantages over traditional gel electrophoresis, such as smaller sample size, automation, and speed. However, current microfluidic chips do not have single nucleotide resolution. In conclusion, the use of denaturing gel electrophoresis to detect altered migration of nonradioactive oligonucleotides provides a useful technique to monitor the catalytic requirements and substrate preference of polynucleotide kinase enzymes.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
We thank Dr. Andrew Sikkema and Andrea Kaminski for their critical reading of this manuscript. This work was supported by the US National Institute of Health Intramural Research Program; US National Institute of Environmental Health Sciences (NIEHS; ZIA ES103247 to R.E.S) and the Canadian Institutes of Health Research (CIHR; 146626 to M.C.P).
Materials
| 0.4 mm 34-well comb | BioRad | 1653848 | |
| 0.4 mm spacer | BioRad | 1653812 | |
| 0.5 M EDTA ph 8.0 | KD Medical | RGF-3130 | |
| 1M Magnesium Chloride | KD Medical | CAC-5290 | |
| 1M Tris pH 8.0 | KD Medical | RGF-3360 | |
| 40% Acrylamide/Bis Solution 29:1 | BioRad | 1610146 | |
| 5M Sodium Chloride | KD Medical | RGF-3720 | |
| ammonium persulfate (APS) | BioRad | 161-0700 | |
| ATP | Sigma | A2383-1G | |
| boric acid | Sigma | B0394 | |
| bromophenol blue sodium salt | Sigma | B5525-5G | |
| Glass Plates | Thomas Scientific | 1188K51 | |
| Hoefer SQ3 Sequencer | Hoefer | N/A | |
| Image J Software | N/A | N/A | https://imagej.nih.gov/ij/ |
| Labeled RNA oligonucleotides | IDT | Custom Order | |
| Pharmacia EPS 3500 Power Supply | Pharmacia | N/A | |
| Steriflip 0. 22 um Filter | Millipore | 5FCP00525 | |
| TEMED | BioRad | 161-0800 | |
| tris base | Sigma | TRIS-RO | |
| Typhoon FLA 9500 gel imager | GE Healthcare | N/A | |
| Ultra Pure DEPC Water | Invitrogen | 750023 | |
| Ultra Pure Glycerol | Invitrogen | 19E1056865 | |
| urea | Fisher Chemical | U15-500 |
Riferimenti
- Pillon, M. C., Stanley, R. E. Nuclease integrated kinase super assemblies (NiKs) and their role in RNA processing. Current Genetics. 64 (1), 183-190 (2018).
- Pillon, M. C., Sobhany, M., Borgnia, M. J., Williams, J. G., Stanley, R. E. Grc3 programs the essential endoribonuclease Las1 for specific RNA cleavage. Proceedings of the National Academy of Sciences U.S.A. 114 (28), E5530-E5538 (2017).
- Gasse, L., Flemming, D., Hurt, E. Coordinated Ribosomal ITS2 RNA Processing by the Las1 Complex Integrating Endonuclease, Polynucleotide Kinase, and Exonuclease Activities. Molecular Cell. 60 (5), 808-815 (2015).
- Dikfidan, A., et al. RNA specificity and regulation of catalysis in the eukaryotic polynucleotide kinase Clp1. Molecular Cell. 54 (6), 975-986 (2014).
- Bernstein, N. K., et al. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Molecular Cell. 17 (5), 657-670 (2005).
- Rio, D. C. 5′-end labeling of RNA with [gamma-32P]ATP and T4 polynucleotide kinase. Cold Spring Harbor Protocols. 2014 (4), 441-443 (2014).
- Paredes, E., Evans, M., Das, S. R. RNA labeling, conjugation and ligation. Methods. 54 (2), 251-259 (2011).
- Hilario, E. End labeling procedures: an overview. Molecular Biotechnology. 28 (1), 77-80 (2004).
- Eastberg, J. H., Pelletier, J., Stoddard, B. L. Recognition of DNA substrates by T4 bacteriophage polynucleotide kinase. Nucleic Acids Research. 32 (2), 653-660 (2004).
- Lillehaug, J. R., Kleppe, K. Kinetics and specificity of T4 polynucleotide kinase. Biochimica. 14 (6), 1221-1225 (1975).
- Richardson, C. C. Phosphorylation of nucleic acid by an enzyme from T4 bacteriophage-infected Escherichia coli. Proceedings of the National Academy of Sciences U.S.A. 54 (1), 158-165 (1965).
- Galburt, E. A., Pelletier, J., Wilson, G., Stoddard, B. L. Structure of a tRNA repair enzyme and molecular biology workhorse: T4 polynucleotide kinase. Structure. 10 (9), 1249-1260 (2002).
- Saito, M., et al. Large-Scale Molecular Evolutionary Analysis Uncovers a Variety of Polynucleotide Kinase Clp1 Family Proteins in the Three Domains of Life. Genome Biology Evolution. 11 (10), 2713-2726 (2019).
- Jain, R., Shuman, S. Characterization of a thermostable archaeal polynucleotide kinase homologous to human Clp1. RNA. 15 (5), 923-931 (2009).
- Weitzer, S., Hanada, T., Penninger, J. M., Martinez, J. CLP1 as a novel player in linking tRNA splicing to neurodegenerative disorders. Wiley Interdisciplinary Reviews RNA. 6 (1), 47-63 (2015).
- Wang, L. K., Lima, C. D., Shuman, S. Structure and mechanism of T4 polynucleotide kinase: an RNA repair enzyme. EMBO Journal. 21 (14), 3873-3880 (2002).
- Zhang, Y., Zhao, J., Chen, S., Li, S., Zhao, S. A novel microchip electrophoresis laser induced fluorescence detection method for the assay of T4 polynucleotide kinase activity and inhibitors. Talanta. 202, 317-322 (2019).
- Cui, L., Li, Y., Lu, M., Tang, B., Zhang, C. Y. An ultrasensitive electrochemical biosensor for polynucleotide kinase assay based on gold nanoparticle-mediated lambda exonuclease cleavage-induced signal amplification. Biosensors and Bioelectronics. 99, 1-7 (2018).
- Wang, L. J., Zhang, Q., Tang, B., Zhang, C. Y. Single-Molecule Detection of Polynucleotide Kinase Based on Phosphorylation-Directed Recovery of Fluorescence Quenched by Au Nanoparticles. Analytical Chemistry. 89 (13), 7255-7261 (2017).
- Liu, H., Ma, C., Wang, J., Chen, H., Wang, K. Label-free colorimetric assay for T4 polynucleotide kinase/phosphatase activity and its inhibitors based on G-quadruplex/hemin DNAzyme. Analytical Biochemistry. 517, 18-21 (2017).
- Du, J., Xu, Q., Lu, X., Zhang, C. Y. A label-free bioluminescent sensor for real-time monitoring polynucleotide kinase activity. Analytical Chemistry. 86 (16), 8481-8488 (2014).
- Jiang, C., Yan, C., Jiang, J., Yu, R. Colorimetric assay for T4 polynucleotide kinase activity based on the horseradish peroxidase-mimicking DNAzyme combined with lambda exonuclease cleavage. Analytica Chimica Acta. 766, 88-93 (2013).
- Castle, C. D., et al. Las1 interacts with Grc3 polynucleotide kinase and is required for ribosome synthesis in Saccharomyces cerevisiae. Nucleic Acids Research. 41 (2), 1135-1150 (2013).
- Pillon, M. C., Sobhany, M., Stanley, R. E. Characterization of the molecular crosstalk within the essential Grc3/Las1 pre-rRNA processing complex. RNA. 24 (5), 721-738 (2018).
- Geerlings, T. H., Vos, J. C., Raue, H. A. The final step in the formation of 25S rRNA in Saccharomyces cerevisiae is performed by 5′–>3′ exonucleases. RNA. 6 (12), 1698-1703 (2000).
- Pillon, M. C., et al. Cryo-EM reveals active site coordination within a multienzyme pre-rRNA processing complex. Nature Structural Molecular Biology. 26 (9), 830-839 (2019).
- Solomatin, S., Herschlag, D. Methods of site-specific labeling of RNA with fluorescent dyes. Methods in Enzymology. 469, 47-68 (2009).
- Giusti, W. G., Adriano, T. Synthesis and characterization of 5′-fluorescent-dye-labeled oligonucleotides. PCR Methods Application. 2 (3), 223-227 (1993).
- Petrov, A., Tsa, A., Puglisi, J. D. Analysis of RNA by analytical polyacrylamide gel electrophoresis. Methods in Enzymology. 530, 301-313 (2013).
