1. Protein preparation
NOTE: The expression and purification of TmPPase has been described elsewhere13.
- Prepare 10 mL of the reactivation buffer solution containing 20 mM 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.5, 3.5% (v/v) glycerol, 2 mM dithiothreitol (DTT), and 0.05% dodecyl maltoside (DDM).
- Prepare 10 mL of the reaction mixture containing 200 mM Tris-Cl pH 8.0, 8.0 mM MgCl2, 333 mM KCl, and 67 mM NaCl.
NOTE: Mg2+ is required to chelate the pyrophosphate as the substrate of mPPase, K+ is required to increase the enzyme activity as TmPPase is a potassium dependent mPPase, and Na+ is needed for the enzyme activity during sodium ion translocation by TmPPase. - Prepare 30 mg/mL liposomes for enzyme reactivation.
- Add 10 mL of 20 mM Tris-HCl pH 8.0 with 1 mM DTT to 0.3 g of L-α-phosphatidylcholine from soybean to 10 mL of 20 mM Tris-HCl pH 8.0 with 1 mM DTT.
- Put the liposome on ice, and sonicate with 1 second pulse interval for 1 minute, pause for 1 minute, and repeat until the solution becomes transparent yellow.
- Aliquot the liposomes, freeze in liquid nitrogen and store at -80 °C until used.
- Reactivate the enzyme.
- Mix 40 µL of the liposomes solution with 22.5 µL of 20% DDM.
- Heat the mixture at 55 °C for 15 min and allow it to cool to room temperature.
- Add 36.5 µL of the reactivation buffer solution, mix, and add 1 µL of concentrated protein (13 mg/mL) to make a total concentration of 0.13 mg/mL.
NOTE: Protein is usually frozen in 10 µL aliquots after purification and thawed on ice before use.
- Take 20 µL of the reactivated enzyme and add to 1,480 µL of the reaction mixture, then mix gently.
NOTE: The addition of the reactivated enzyme to the reaction mixture should be performed just before it is used.
2. Compound preparation
- Dissolve the compounds in dimethyl sulfoxide (DMSO) to make stock solutions of 25−100 mM in 50−200 µL, based on the availability of the compounds.
NOTE: All compounds used here (Figure 2A) have been published previously9. If the compound solubility is low, the stock concentration can be adjusted accordingly. - Prepare three different concentrations of each compound in water.
NOTE: The final concentrations in the reaction mixture will be 1, 5, and 50 micromolar or 1, 5, and 20 micromolar for soluble and sparingly soluble compounds, respectively.- Dilute the stock solution with water to 1 mL in microtubes to give 2 µM, 10 µM and 100 µM for soluble compounds, or alternatively 2 µM, 10 µM and 40 µM for sparingly soluble compounds.
- Vortex the compound solution instantly after dilution of the stock solution for proper mixing.
- Check for compound aggregation using a nephelometer.
NOTE: This was studied as triplicates in three concentrations (1 µM, 5 µM and 20 µM) and normalized to the blank in a 96 well plate.- Dispense 75 µL of the reaction mixture into each well using a multichannel pipette.
- Add 75 µL of each compound (for the blank, use 75 µL of water instead) and mix by pipetting up and down 5×.
- Measure each well at 300 V using a microplate nephelometer.
3. Reagents for the assay preparation
- Prepare the arsenite-citrate solution.
- Weigh 5 g of sodium arsenite and 5 g of trisodium citrate dihydrate.
CAUTION: Sodium arsenite is toxic, thus use proper protective equipment and handle with special care. As precaution, do not handle before all necessary safety precautions have been read and understood. Handle only in a fume hood in order not to inhale dust/vapors of the compound or its solution(s). If inhaled, move to fresh air and obtain medical attention. Wear appropriate chemical safety goggles, protective gloves and clothing to avoid ingestion and eye/skin contact. If swallowed, call immediately a poison center or doctor/physician. If it gets on the skin or in the eye(s), wash with plenty of water and obtain medical attention. - Dissolve into 100 mL of water.
- Add 5 mL of glacial acetic acid, mix, and add water to 250 mL.
- Store at room temperature protected from light.
NOTE: The solution is stable for more than a year.
- Weigh 5 g of sodium arsenite and 5 g of trisodium citrate dihydrate.
- Prepare solution A and solution B.
- For solution A, add 10 mL of ice cold 0.5 M HCl to 0.3 g of ascorbic acid. Dissolve the ascorbic acid by vortexing.
- For solution B, add 1 mL of ice cold water to 70 mg of ammonium heptamolybdate tetrahydrate and vortex to dissolve.
NOTE: Store both solutions on ice until use. For the consistency of the assay result, both solutions can be stored on ice for a maximum of one week.
- Prepare the phosphate (Pi) standard with the concentration of 0 µM, 62.5 µM, 250 µM and 500 µM for calibration.
- Add 0 µL, 25 µL, 50 µL, and 100 µL of 5 mM Na2HPO4 dihydrate to four microtubes containing 370 µL of the reaction mixture.
- Top up to 1 mL with water.
4. Activity assay for one 96 well plate
NOTE: See Figure 1 for the schematic workflow of the assay.
- Add 1 mL of solution B to 10 mL of solution A, mix by vortexing and store the solution on ice.
NOTE: This solution should be transparent and yellow. Keep solution A + B on ice for at least 30 min prior to use. However, use the solution within 3 h as it will go bad after long-term storage. - Add 40 µL of 0 µM, 62.5 µM, 250 µM and 500 µM Pi standard to the tube strips in triplicate using a multichannel pipette.
NOTE: The reaction mixture with no Pi added will be used as a blank. - Add 25 µL of compound solution to the tube strips using a multichannel pipette.
NOTE: Each compound has three different concentrations in triplicate which is enough for initial estimation of the half maximal inhibitory concentration (IC50). For a more accurate IC50 determination, eight different compound concentrations can be used. For the uninhibited enzyme the compound solution is replaced with equal amount of water. As positive controls 2.5 µM, 25 µM, and 250 µM of imidodiphosphate (IDP) sodium salt were used. - Add 15 µL of mPPase solution mixture to the tube strips (except to the tubes containing Pi standard) using a multichannel pipette.
- Seal the tube strips with an adhesive sealing sheet. Cut the sealing sheet to separate each tube strip.
- Pre-incubate the samples for 5 min at 71 °C. Place the samples on the heating block with 20 s interval between each strip in order to minimize the time consumption during the subsequent steps.
- For each strip, open the adhesive sealing. Add 10 µL of 2 mM sodium pyrophosphate dibasic using a multichannel pipette and mix by pipetting up and down for 5×. Seal the tube strip again using the same sealing.
NOTE: This step might initially be difficult to accomplish in 20 s; however, it will become easier after some assays. - Incubate at 71 °C for 5 min.
- Place the samples on the cooling apparatus with 20 s interval between each strip. Let them cool for 10 min but centrifuge each strip briefly after 5 min of cooling, to decant water drops under the sealing sheet, then put it back to the cooling apparatus and remove the sealing.
NOTE: The cooling apparatus can simply be made by placing a 96 well PCR plate on a polystyrene Petri dish (size 150 mm × 15 mm) filled with water and frozen for at least 1 h. The apparatus should be taken out from the freezer about 5 min prior to the beginning of the assay. Do not take out the cooling apparatus right before sample cooling as it will freeze the reaction mixture and hinder color development. - After 10 min of cooling, add 60 µL of solution A + B, mix by pipetting up and down for 5× and keep the tube strips on the cooling apparatus for 10 min.
- Add 90 µL of the arsenite-citrate solution and keep at room temperature for at least 30 min to produce a stable blue color.
CAUTION: Due to its toxicity all solutions containing sodium arsenite should be handled with extra care at all time. Thus, the addition of arsenite-citrate solution should be done in a fume hood. - Dispense 180 µL of each reaction mixture into a clear 96 well polystyrene microplate.
- Measure the absorbance of each well at 860 nm using a microplate spectrophotometer.
5. Result analysis
- Average the triplicates of each sample and the Pi standards. Then subtract with the blank to eliminate the background signal.
- Make a calibration curve by plotting the absorbance (A860) values against the amount of Pi standard (nmol) and perform a linear regression to obtain the trendline function using the following formula:
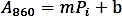
- Calculate the phosphate amount (nmol) released from the enzymatic reaction based on the linear regression formula above.
- Calculate the specific activity using the following formula:
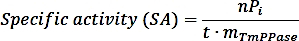
where nPi is the amount of phosphate released from the reaction (nmol), t is the reaction time (min), and mTmPPase is the amount of the pure TmPPase used in the assay (mg). - Calculate the percent activity for each inhibitor concentration using the following formula:
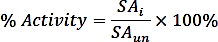
where SAi is the specific activity of a sample with inhibitor and SAun is the specific activity of the uninhibited sample. - Calculate the logIC50 (estimate) and IC50 (estimate) with a nonlinear regression fit from the four-parameter dose-response curve using the following formula:
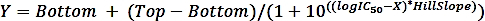
where X is log of concentration ( µM), Y is activity (%), Top and Bottom are plateaus in the same unit as Y (100% and 0%, respectively), logIC50 has the same log units as X, and HillSlope = slope factor or hill slope, which is unitless.
NOTE: Software (Table of Materials) is used for the fitting. Use the concentration of 0.01 µM (instead of 0.00 µM) for the sample without inhibitor as the logarithm of zero is not defined.
In this protocol, eight compounds (1−8) were tested (Figure 2A) together with IDP, a common inhibitor of pyrophosphatases, as a positive control. Each compound was tested at three different concentrations (1 µM, 5 µM and 20 µM) in triplicate. The workflow of the screening is depicted in Figure 1, starting from sample and reagent preparation until the absorbance measurement at 860 nm.
At the end of this protocol, after the addition of solution A + B and arsenite-citrate, the solutions develop a stable blue color with the maximum absorption at 709 nm and 860 nm14 due to the complex formation of phosphate ions with molybdate that can be observed and shows the occurrence of the enzymatic reaction. For this experiment, we use the absorbance at 860 nm for the measurement of Pi amount released as it has better detection limit and sensitivity compared to the absorbance at 709 nm15. The blue color is fully developed in 30 min of incubation at room temperature and stable for at least 5 h14. The assay has the sensitivity down to Pi concentration of 10 µM and the absorbance is linear over a concentration range of 10−800 µM14. In the representative result here, wells E1−E3 (Figure 2C) contain the reaction mixture without inhibitor and the blue solution can be observed at the end of the assay. This can also be observed at low compound concentrations where complete inhibition has not been reached, as in wells F1−F3 for IDP and wells A4−A6 for compound 1 (ATC, a recently known uncompetitive inhibitor of TmPPase9) at the concentration of 2.5 µM and 1 µM, respectively. The higher concentration of IDP and compound 1, the less to no blue color can be observed (G1−G3 and H1−H3 for IDP and B4−B6 and C4−C6 for compound 1) indicating inhibition of the enzymatic activity. All three concentrations of non-inhibiting compounds (2, 3, and 8) displayed the same blue color intensity as wells E1−E3 without any inhibitor (Figure 2C).
After the absorbance measurement at 860 nm, the data can be processed and analyzed (see protocol section 5). Figure 2D shows the calibration plot of Pi standard with its linear fitting (y = 0.0576x + 0.0019; r2 = 0.999). Figure 3 shows the plot of enzymatic activity (%) against the concentration of each tested compound. For compounds with inhibition activity, a nonlinear curve fitting is also shown. IDP, used as a positive control, clearly shows a decrease in activity at higher concentration. The IC50 (estimate) calculated based on three different concentrations is 88.2 µM (Table 1), which is similar to the previous measurement (80.0 µM) with eight concentration points14. Compounds 1, 4, 5, 6, and 7 showed a similar trend as IDP since the concentration increased with the IC50 (estimate) of approximately 1.3 µM, 7.4 µM, 19.0 µM, 37.4 µM, and 156.1 µM, respectively (Table 1). For compounds 2, 3, and 8 no reduction in activity or inhibition can be observed at the assay concentrations. An additional assay with eight concentration points can be done to generate precise IC50. Figure 4 shows the inhibition curve for compounds 1, 5, 6, 7 and 8 with an IC50 of 1.7 µM, 21.4 µM, 58.8 µM, 239.0 µM and >500 µM, respectively9.
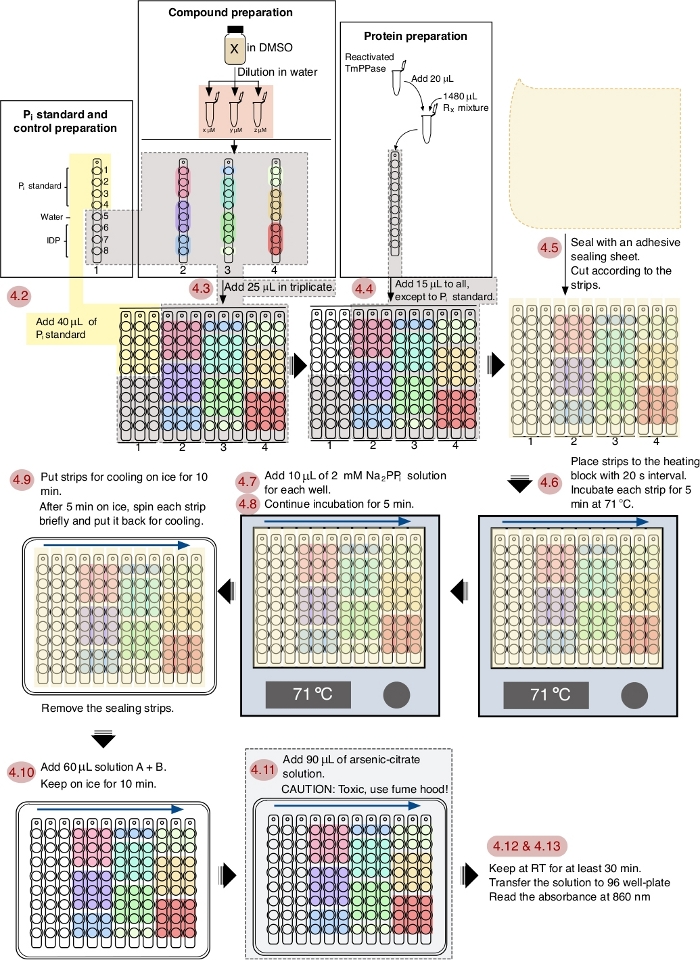
Figure 1: A schematic workflow of TmPPase inhibition assay in a 96 well plate format. The red numbering shows the steps of the assay according to the protocol and the blue arrows show the interval order. Please click here to view a larger version of this figure.
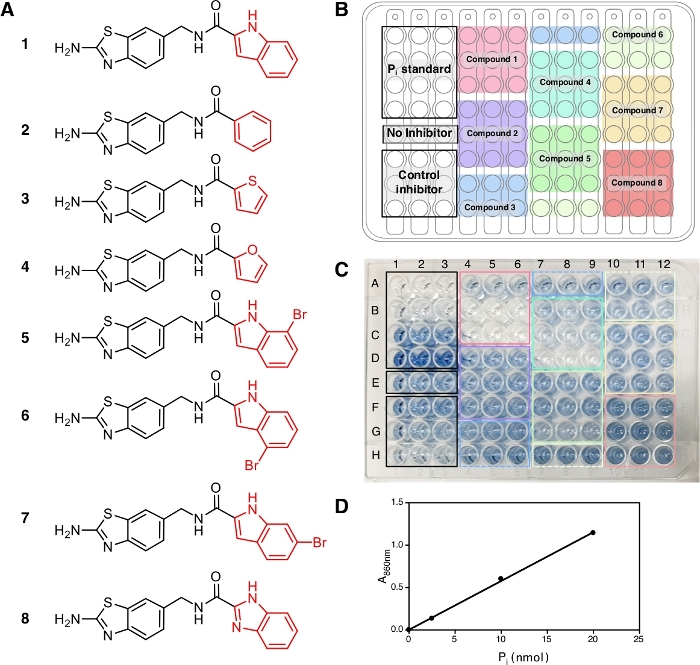
Figure 2: Samples, their arrangement and color development in a 96 well plate. (A) The structures of compounds 1−8 used for the assay. The inhibition activity of these compounds has been reported in Vidilaseris et al.9. (B) Sample arrangement. (C) Color development, 30 min after the addition of arsenite-citrate solution. The concentrations of control inhibitor (IDP) and samples used, arranged from the top to the bottom, are 2.5 µM, 25 µM, and 250 µM concentration and 1 µM, 5 µM, and 20 µM concentration, respectively. The intensity of the blue color corresponds to the amount of Pi released due to the enzymatic reaction and the lack of color corresponds to no enzymatic reaction. (D) Calibration curve for Pi standard (nmol) against A860 with linear fitting (y = 0.0576x + 0.0019; r2 = 0.999). Please click here to view a larger version of this figure.
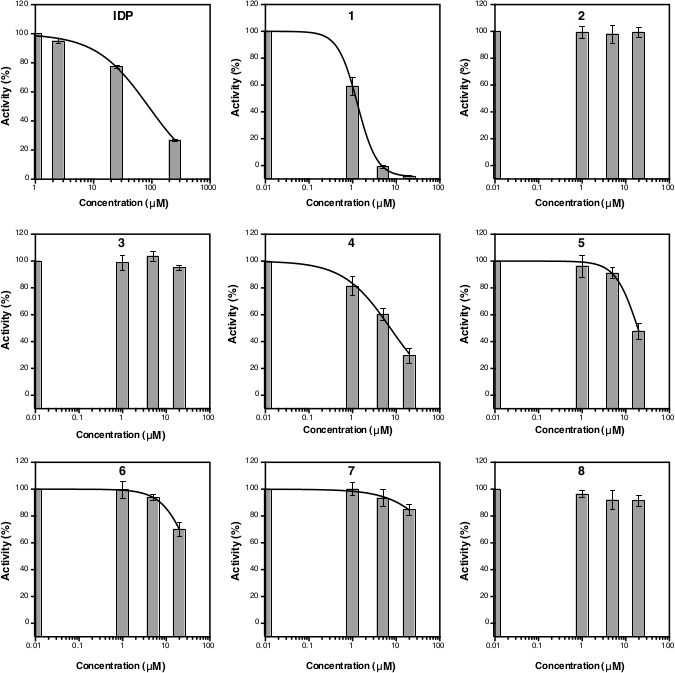
Figure 3: Curve of the TmPPase percent activity for three different inhibitor concentrations. The nonlinear regression curves to calculate the IC50 (estimate) are shown for IDP as well as for compounds 1, 4, 5, 6 and 7 but not for compounds 2, 3, and 8 as they were not inhibiting TmPPase activity at the assay concentrations. The logIC50 and IC50 (estimate) of each compound is shown in Table 1. All data are shown as mean ± SD with three replicates. Please click here to view a larger version of this figure.
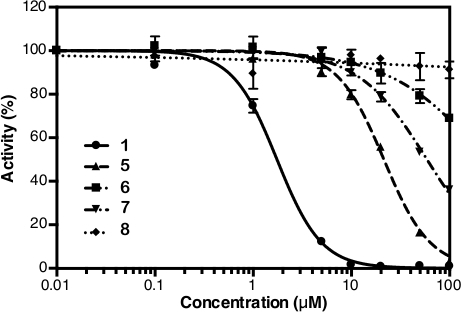
Figure 4: Inhibition curve from eight concentration points of compounds 1, 5, 6, 7 and 8. This figure is taken from Vidilaseris et al.9 with slight modification. All data are shown as mean ± SD with three replicates. Please click here to view a larger version of this figure.
| Sample | LogIC50 | IC50 (estimate) (µM) |
| IDP | 1.95 ± 0.0142 | 87.9 ± 2.46 |
| 1 | 0.112 ± 0.0274 | 1.29 ± 0.0816 |
| 2 | − | no inhibition |
| 3 | − | no inhibition |
| 4 | 0.870 ± 0.0447 | 7.39 ± 0.760 |
| 5 | 1.28 ± 0.0296 | 19.0 ± 1.29 |
| 6 | 1.57 ± 0.0846 | 37.4 ± 7.29 |
| 7 | 2.19 ± 0.366 | 156 ± 131 |
| 8 | − | no inhibition |
Table 1: LogIC50 and IC50 (estimate) of IDP and compounds 1−8 based on the data from Figure 3.
