All experiments with mice were approved by the Institutional Animal Care and Use Committee (IACUC) at Yale University (Protocol 2021-11117) and performed in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Animal care and housing complied with the Guide for the Care and Use of Laboratory Animals33 and were provided by the Yale Animal Resource Center (YARC). Animals were maintained in a 12 h light/dark cycle with ad libitum access to food and water. Five to eight mice or two to four rats per genotype or condition are required for the following protocol. Fewer rats are necessary due to their larger brain volumes. Similarly, the age of the experimental animals may affect fraction yield; additional mice may be required for ages less than 2 months. Otherwise, the outlined procedures apply to both murine species and healthy adult animals of any age. The representative data presented in this study utilized wild-type (C57BL/6J) mice (age = 2 months; four males and four females per replicate) obtained from a commercial source (see Table of Materials).
1. Experimental preparation
NOTE: This protocol requires ~11 h for a single researcher to complete. It is highly recommended to complete benchtop setup (Figure 1), buffer preparation (Table 1), the precooling of centrifuges and rotors to 4 °C, and the collection and labeling of necessary materials and equipment (see Table of Materials) the day prior to protocol execution, where applicable.
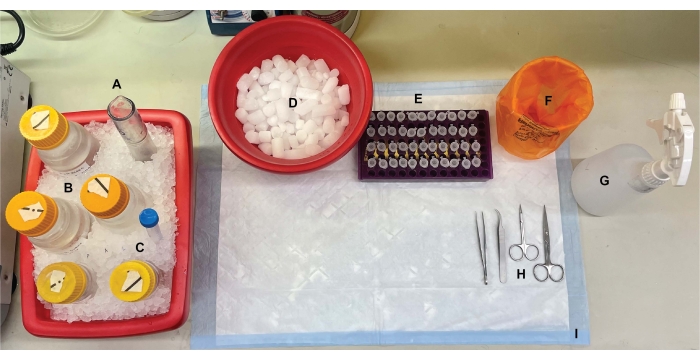
Figure 1: Benchtop setup. Prior to brain dissections, (A) Dounce glass homogenizers and (B) all buffers were chilled on ice. (C) Protease inhibitor stock solutions were thawed on ice. A second container of wet ice for centrifuge tubes, a Dewar of liquid nitrogen (not shown), and (D) a container of dry ice for short-term storage of the samples flash-frozen in liquid nitrogen were obtained. (E) Microcentrifuge tubes were pre-labeled for all samples, as four aliquots of each subcellular fraction sample per genotype or condition were collected during this procedure (time-saving tip: thoroughly label all the tubes the day before the experiment is performed). (F) An appropriate biohazard waste container, (G) 70% ethanol, (H) surgical tools, and (I) an absorbent surface pad. The required centrifuge tubes and disposables were set aside for efficient access during protocol implementation (not shown). Please click here to view a larger version of this figure.
- Prepare the benchtop for surgery and collect the scissors and forceps required for brain excision (see Table of Materials). Pre-label 1.5 mL microcentrifuge tubes for mouse tail biopsies and four tubes per collected fraction, as outlined in Figure 2.
- Obtain two containers of wet ice, one container of dry ice, and a benchtop liquid nitrogen Dewar flask.
- Thaw phenylmethylsulfonyl fluoride (PMSF), pepstatin A, aprotinin, and leupeptin stock solutions on ice (see Table of Materials). Prepare the necessary buffers (Table 1).
NOTE: Sucrose solutions can be prepared in advance and stored at 4 °C. However, protease inhibitors (thawed stocks and tablets) must be added fresh to all the buffers at the start of the experiment due to the instability of these reagents in aqueous solutions. Further, all the buffers must be prepared with detergent-free glassware and detergent-free water to enable the collection of intact synaptosomes. - Chill all the buffers and glass Dounce homogenizers (see Table of Materials) on ice. Set the centrifuges to 4 °C and chill the rotors to 4 °C.
- Add 14 mL of Buffer A (Table 1) to a Dounce homogenizer on ice.
Table 1: Composition of the subcellular fractionation buffers. Please click here to download this Table.
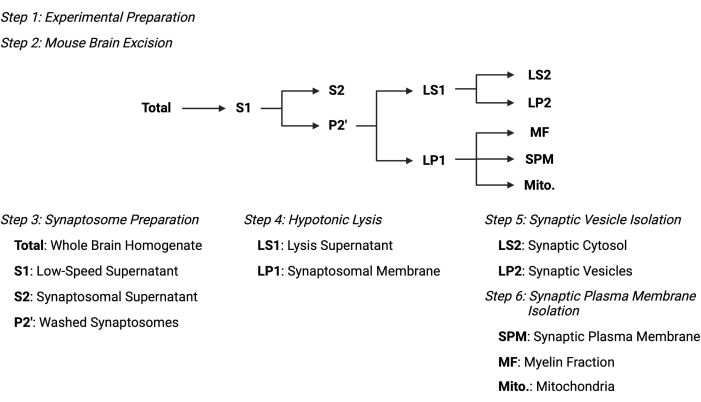
Figure 2: Overview of the subcellular fractionation protocol. Summary schematic of the subcellular fractionation steps and collected samples. Please click here to view a larger version of this figure.
2. Mouse brain excision
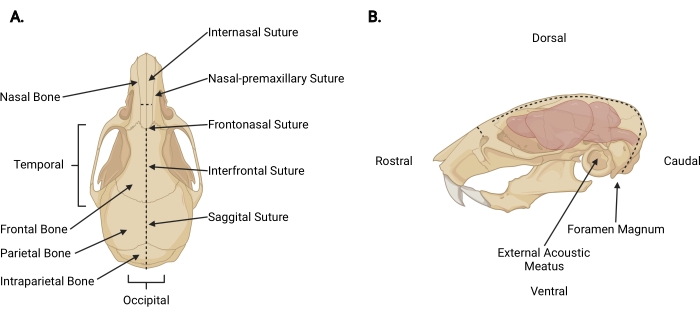
Figure 3: Craniofacial anatomy. (A) Dorsal view of a mouse skull with relevant cranial structures indicated. (B) Left lateral view of a mouse skull and brain with relevant cranial structures and anatomical directions indicated. The dashed lines represent the locations where incisions should be made. Please click here to view a larger version of this figure.
- Deeply anesthetize each mouse with 100% isofluorane in an anesthesia chamber located in a fume hood or biosafety cabinet using an open drop method34. Sacrifice each mouse by cervical spine dislocation followed swiftly by decapitation. Alternate between genotypes or experimental groups for each sacrifice and dissection12. Obtain tail biopsies after euthanasia by excising 2 mm of the distal tail tip with fine scissors. Store the tissue for genotyping.
- Spray the decapitated head with 70% ethanol to prevent hair from adhering to the tissue and surgical instruments during dissection.
- Insert fine scissors under the skin at the decapitation incision to a pericranial depth and make a midsagittal incision up to the internasal suture (Figure 3A) to retract the scalp from the skull.
- Working from the occipital area toward each temporal aspect, trim the fascia and muscle to expose the external surface of the skull beyond each external acoustic meatus (Figure 3B).
- Secure the scalp and rostral aspect of the skull with the non-dominant hand. With the other, insert fine scissors 2 mm into the caudal side of the foramen magnum, where the spinal cord is visible exiting. Make a midline incision until the scissors reach the internal surface of the intraparietal bone (Figure 3; dashed lines).
NOTE: During the initial incision, the scissors must be parallel to the spinal cord with pressure applied toward the internal surface of the skull to prevent damage to the brainstem and cerebellum. - Change the angle of the scissors so the blades run parallel with the dorsal surface of the skull. Continue advancing the midsagittal incision rostrally through the parietal and frontal bones, using the sagittal and interfrontal sutures as a guide. Use constant upward pressure to avoid damage to the cortex. Terminate the incision just beyond the internasal suture (Figure 3A).
- Make a small perpendicular incision (~3 mm) to the nasal bone, rostral to the internasal suture, by placing the scissors perpendicular to the skull with each blade positioned at a nasal-premaxillary suture and making one even cut (Figure 3; dashed lines).
NOTE: This step will increase the ease of retracting the skull and will be critical for collecting the olfactory bulb if this area is of interest. - While securing the rostral aspect, use one side of a pair of textured forceps to gently lift the skull up from the brain, then laterally and ventrally. Repeat along the midline as needed, then for the other hemisphere until the entire brain surface is exposed.
- Using curved forceps or a fine spatula, gently lift the rostral side of the brain. Cut the optic and cranial nerves to complete the excision from the skull.
- For each condition, collect five to eight mouse brains together into the chilled glass Dounce homogenizer containing 14 mL of Buffer A (Table 1).
3. Synaptosome preparation
NOTE: The schematics of this procedure are shown in Figure 4.
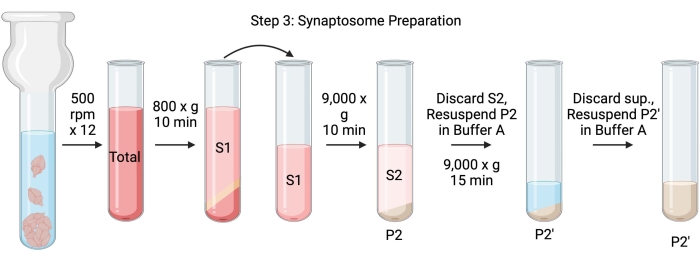
Figure 4: Synaptosome preparation. Schematic of step 3, the generation of synaptosomes (P2'). Please click here to view a larger version of this figure.
- Homogenize the brains using a glass Dounce homogenizer in 12 up-down passes at 500 rpm (total). Pause briefly at each downstroke to ensure thorough homogenization of the tissue. Homogenize preferentially in an ice bath to avoid warming and protein denaturation. Take 5 µL aliquots for protein concentration determination by the bicinchoninic acid assay (BCA, see Table of Materials). Take 100 µL of whole brain lysate aliquots for western blot (WB). For this and all subsequent samples (Figure 2), take two aliquots for BCA and two aliquots for WB. Flash-freeze all the collected aliquots in liquid nitrogen and store them at −80 °C.
- Spin the total brain homogenate in a high-speed round bottom centrifuge tube (14 mL) (see Table of Materials) at 800 x g for 10 min at 4 °C to obtain the supernatant (S1). Transfer S1 to a new centrifuge tube, leaving the pellet behind (P1), which contains intact cells and nuclei. Avoid pipetting up the fluffy, white, loose, superficial pellet. Take 2 x 5 µL of S1 for BCA and 2 x 100 µL of S1 for WB.
- Spin S1 at 9,000 x g for 15 min at 4 °C to obtain the synaptosomal supernatant (S2) and crude synaptosome pellet (P2). Take 2 x 10 µL of S2 for BCA and 2 x 500 µL of S2 for WB. Discard the supernatant after obtaining aliquots and proceed to the next step with the pellet.
- Resuspend P2 in 3 mL of ice-cold Buffer A with protease inhibitors and centrifuge at 9,000 x g for 15 min at 4 °C to obtain the supernatant (S2') and washed synaptosomes (P2'). Discard the supernatant and keep the pellet.
- Resuspend P2' in 3 mL of Buffer A. Avoid resuspending the dark red portion at the bottom of the pellet, which mainly contains mitochondria. Take 2 x 20 µL of P2' for BCA and 2 x 100 µL of P2' for WB.
NOTE: This can be achieved by gently pipette-mixing the edges and surface of the pellet to resuspend the white washed synaptosomes while directing the pipette tip away from the red center of the pellet.
4. Hypotonic lysis
NOTE: The schematics of this procedure are shown in Figure 5.
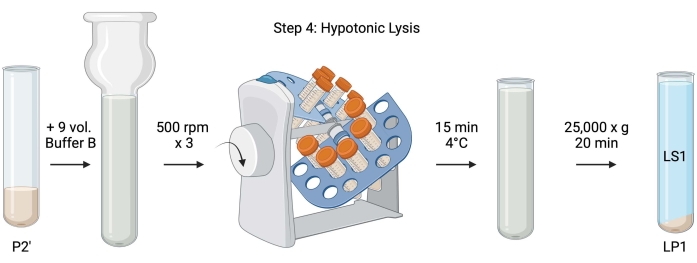
Figure 5: Hypotonic lysis. Schematic of step 4, the hypotonic lysis of synaptosomes to generate the lysis supernatant (LS1) and synaptosomal membrane fractions (LP1). Please click here to view a larger version of this figure.
- For the hypotonic lysis of washed synaptosomes, add 9 volumes of chilled Buffer B (Table 1) to resuspended P2' (~27 mL). Homogenize the synaptosomes in a glass Dounce homogenizer (three up-down passes at 500 rpm).
- Transfer the samples to 50 mL capped conical centrifuge tubes. Rotate them on a tube revolver in a 4 °C cold room for 15 min.
- Centrifuge lysed P2' at 25,000 x g for 20 min at 4 °C to obtain the lysis supernatant (LS1) and the lysis pellet containing synaptosomal membranes (LP1). Take 2 x 50 µL of LS1 for BCA and 2 x 400 µL of LS1 for WB. Transfer LS1 into a capped centrifuge tube for ultracentrifugation (see Table of Materials).
5. Synaptic vesicle isolation
NOTE: The schematics of this procedure are shown in Figure 6.
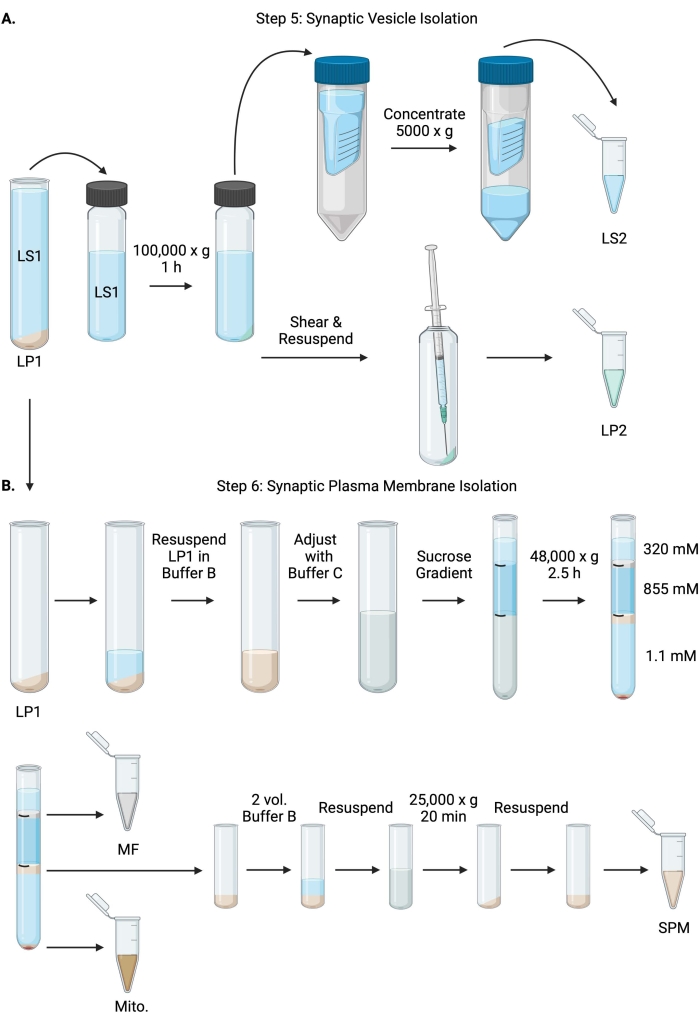
Figure 6: Synaptic vesicle isolation and synaptic plasma membrane isolation. (A) Schematic of step 5, the isolation of synaptic cytosol (LS2) and synaptic vesicle (LP2) fractions, and (B) step 6, the generation of myelin (MF), synaptic plasma membrane (SPM), and mitochondrial (Mito.) fractions following the ultracentrifugation of sucrose gradients. Please click here to view a larger version of this figure.
- Centrifuge LS1 in a fixed angle ultracentrifuge rotor (see Table of Materials) at 100,000 x g for 60 min at 4 °C to obtain synaptic cytosol supernatant (LS2) and synaptic vesicle pellet (LP2). LP2 will be small, translucent, and strongly adhered to the side of the centrifuge tube.
- Resuspend LP2 in 500 µL of Buffer A. Using a 23 G needle and a 1 mL syringe, shear LP2 with gentle trituration. Take 2 x 10 µL of LP2 for BCA and 2 x 250 µL of LP2 for WB.
- Transfer LS2 (~30 mL) to centrifugal filter units with a 10 kDa cutoff (see Table of Materials).
NOTE: If proteins smaller than 10 kDa are of interest, 4 kDa cutoff centrifugal filter units are available but will result in longer spin times. - Concentrate LS2 to approximately 0.5 mL by spinning at 5000 x g for up to 1 h at 4 °C. Take 2 x 10 µL of concentrated LS2 for BCA and 2 x 250 µL of concentrated LS2 for WB. After starting the spin, proceed directly to step 6.1.
6. Synaptic plasma membrane isolation
- Resuspend LP1 (step 4.3) in 1 mL of Buffer B (Table 1). Take 2 x 10 µL of LP1 for BCA and 2 x 50 µL of LP1 for WB. Adjust the remaining LP1 to a final volume of 7.5 mL and a final sucrose concentration of 1.1 M with Buffer B and Buffer C (Table 1).
- Transfer 7.5 mL of resuspended LP1 into a 14 mL ultracentrifuge tube (see Table of Materials). Carefully overlay LP1 with 3.75 mL of Buffer D (Table 1), and then overlay with 1.25 mL of Buffer A (or a larger volume to fill just below the top of the centrifuge tube). Avoid pipetting down the side of the tube, which will disrupt the sucrose gradient interfaces. After overlaying each sucrose fraction, mark the top of the solution with a pen. Balance the tubes for ultracentrifugation by weight, not volume, with the dropwise addition of Buffer A to within 10 mg. Centrifuge at 48,000 x g for 2.5 h at 4 °C in a swinging bucket ultracentrifuge rotor (see Table of Materials).
- Acquire images of the intact gradients following ultracentrifugation to document the distinctness of each sucrose interface and the success of fractionation.
- Carefully remove the superficial layer of 320 mM sucrose (Buffer A). Recover the myelin fraction (MF) at the 320 mM/855 mM sucrose interface in an 800 µL volume. Recover the synaptic plasma membrane (SPM) fraction at the 855 mM/1.1 M sucrose interface in a 1,000 µL volume. Pipette each fraction up from the wall of the tube in a circular manner to ensure the complete fraction is collected. Carefully aspirate off the remaining sucrose and recover the mitochondrial pellet (Mito.) by resuspending in 200 µL of Buffer B. Take 2 x 100 µL of MF for BCA and 2 x 10 µL of Mito. for BCA; divide the remainder of MF and Mito. samples in half for WB.
- Dilute the SPM fraction with 2 volumes of Buffer B (~2 mL), and then centrifuge in a fixed angle rotor in a 3.5 mL centrifuge tube (see Table of Materials) at 25,000 x g for 20 min at 4 °C. Discard the supernatant and resuspend the SPM pellet in Buffer A for a final volume of 250 µL. Take 2 x 5 µL of SPM for BCA and divide the remaining SPM in half for WB.
- Perform a BCA to determine the protein concentration of each sample, accounting for variable aliquot volume.
NOTE: For WB analysis, the suggested working protein concentration for all subcellular fractions is 2 µg/µL (or as high as achievable for LS1 and MF).
The presented method results in 11 brain subcellular fractions that can be subjected to further purification and various forms of downstream analysis35,36. The gold standard method to assess the quality of synaptosomes, SVs23, and other components is electron microscopy (EM) (Figure 7). Quantitative immunoblotting for proteins that are present in specific subcellular fractions can also be performed to assess markers of fraction purity (Figure 8). For example, immunoblot analysis of fractions reveals the enrichment of N-cadherin (CDH2, UniProt name) in the synaptic plasma membrane fraction (SPM), α-synuclein (SYUA) in the synaptic cytosol (LS2), synaptophysin (SYPH) in the synaptic vesicle fraction (LP2), and myelin basic protein (MBP) in the myelin fraction (MF) when compared to protein levels in the initial whole brain homogenate (Total) (Figure 8). Once fraction purity has been established (for example, note the absence of CDH2 in the LS2 fraction or the many-fold increase in SYPH in the LP2 fraction), quantitative immunoblotting can be used to determine the localization of proteins of interest or query differences in protein distribution between genotypes or treatments. Understanding the subcellular localization of synaptic proteins can enable the dissection of previously undescribed protein functions. Further, this method may elucidate trafficking defects or synaptic dysfunction in disease states, especially when paired with functional assays. For example, our team has used this method to identify a pool of enzymatically active palmitoyl protein thioesterase 1 that is enriched in the synaptic cytosol19.
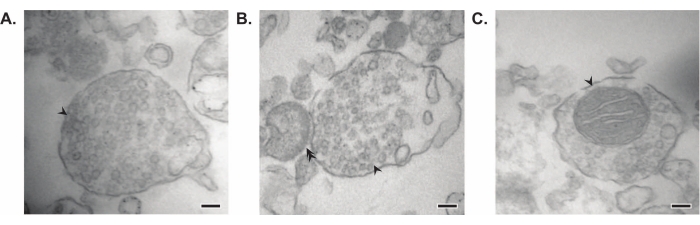
Figure 7: Electron microscopy (EM) of synaptosomes. (A) Representative EM image of a synaptosome containing synaptic vesicles (arrow). (B) Representative EM image of a synaptosome with both pre- (arrow) and post-synaptic components (double arrow). (C) Representative EM image of a synaptosome containing synaptic vesicles and a mitochondrion (arrow) (scale bars = 100 nm). Please click here to view a larger version of this figure.
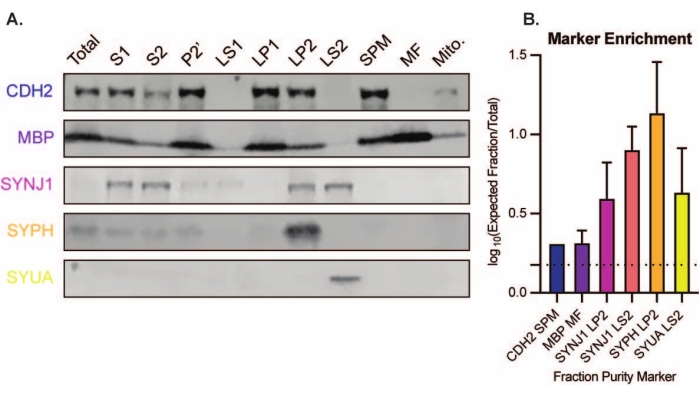
Figure 8: Immunoblot analysis of subcellular fractions. (A) Markers of subcellular fraction purity (indicated with UniProt nomenclature) are appropriately localized compared to the whole brain homogenate (total): N-cadherin (CDH2) in the synaptic plasma membrane fraction (SPM), synaptophysin 1 (SYPH) and synaptojanin 1 (SYNJ1) in the synaptic vesicle enriched fraction (LP2), α-synuclein (SYUA) in the synaptic cytosol (LS2), and myelin basic protein (MBP) in the myelin fraction (MF). (B) Immunoblot quantification analysis reveals the enrichment (fold-change from total) of fraction purity markers. Data are represented as mean ± standard deviation on a log10 scale. The dotted line indicates a 1.5-fold change (y = 0.176) (n = 3 replicate experiments with 8 wild-type mice; age = 2 months; n = 4-5 blots for SYPH, SYUA, MBP, with n = 3 plotted values previously published by Gorenberg et al.19; n = 5 for SYNJ1; n = 1 for CDH2). Please click here to view a larger version of this figure.
