1. Prepare silanized coverslips and microscope slides to prevent adsorption of proteins to chamber surfaces
NOTE: This is a 2-day process. Silanized slides may be prepared up to 1 month in advance of use.
- Place no. 1 coverslips (24 mm x 24 mm) and microscope slides (1 in x 3 in) in a designated rack that will fit in the plasma cleaner. Place rack in plasma cleaner and run for 20 min.
- Transfer coverslips and slides to a new rack designated only for use with silane and place rack in glass container to clean the glasses as described below.
- Immerse coverslips and slides in 100% acetone for 1 h. Immerse coverslips and slides in 100% ethanol for 10 min.
- Immerse coverslips and slides in deionized water (DI) for 5 min. Repeat the cleaning steps two more times.
- Immerse coverslips and slides in freshly prepared 0.1 M KOH for 15 min. Immerse coverslips and slides in fresh DI for 5 min. Repeat this step two more times.
- Air dry coverslips and slides for 10 min. Treat cleaned coverslips and slides with silane to produce hydrophobic surfaces as described below.
NOTE: Complete the following steps in a fume hood.- Immerse dried coverslips and slides in 2% silane (dissolved in toluene) for 5 min. Use a funnel to pour silane back into its designated bottle to reuse up to five times.
- Immerse coverslips and slides in 100% ethanol for 5 min. Replace ethanol with fresh ethanol. Immerse coverslips and slides for 5 min.
- Immerse coverslips and slides in fresh DI for 5 min. Repeat the ethanol and DI wash step two more times using fresh ethanol and DI each time. Air dry coverslips and slides for 10 min.
2. Preparing active actin-microtubule composite driven by myosin mini-filaments
- Remove inactive myosin via actin filament binding and perform pull-down via ultracentrifugation as described below.
- Polymerize actin into filaments. Using a precision micropipette and sterile pipet tips, combine in a microcentrifuge tube: 1.87 µL of DI, 1.3 µL of 10x G-buffer, 1.3 µL of 10x F-buffer, 1.63 µL of 4 M KCl, 4.53 µL of actin (47.6 µM), and 1.08 µL of 100 µM phalloidin.
NOTE: To ensure sufficient polymerization, the actin concentration and actin:phalloidin molar ratio should be 18.4 µM and 2:1, respectively. - Gently pipet the solution up and down to mix and then set on ice in the dark for ≥1 h. Cool ultracentrifuge to 4 °C. Remove myosin aliquot from -80 °C and put on ice.
NOTE: Complete step 2.2 at this point while actin is polymerizing. - After ≥1 h of actin polymerization, add 1.3 µL of 10 mM ATP and 2 µL of 19 µM myosin to the polymerized actin.
NOTE: The actin:myosin molar ratio should be >5 to ensure sufficient removal of inactive myosin motors (i.e., dead heads). - Gently pipet the solution up and down to mix. Transfer into an ultracentrifuge grade tube.
Centrifuge at 4 °C and 121,968 x g for 30 min.
- Polymerize actin into filaments. Using a precision micropipette and sterile pipet tips, combine in a microcentrifuge tube: 1.87 µL of DI, 1.3 µL of 10x G-buffer, 1.3 µL of 10x F-buffer, 1.63 µL of 4 M KCl, 4.53 µL of actin (47.6 µM), and 1.08 µL of 100 µM phalloidin.
- Prepare co-entangled composite network of actin filaments and microtubules as described below.
NOTE: Begin 30 min before myosin spin-down (step 2.1.4).- Set a heat block to 37 °C. Use a precision micropipette and sterile pipet tips to add the following to a microcentrifuge tube: 13.9 µL of PEM, 3 µL of 1% Tween20, 1.55 µL of 47.6 µM actin, 0.36 µL of 34.8 µM R-actin, 0.3 µL of 250 mM ATP, 0.87 µL of 100 µM phalloidin, 1.91 µL of 5-488-tubulin, 0.3 µL of 100 mM GTP, and 0.75 µL of 200 µM Taxol, to a total volume of 23 µL.
NOTE: The concentrations of actin and tubulin listed are for a composite with 2.9 µM actin and 2.9 µM tubulin. Total protein concentration is c = cA + cT = 5.8 μM and molar actin fraction is cA/(cA + cT) = ΦA = 0.5. See step 2.5 to adjust these values. - Gently pipet the solution up and down to mix and place on a 37 °C heat block protected from light for 1 h.
- Set a heat block to 37 °C. Use a precision micropipette and sterile pipet tips to add the following to a microcentrifuge tube: 13.9 µL of PEM, 3 µL of 1% Tween20, 1.55 µL of 47.6 µM actin, 0.36 µL of 34.8 µM R-actin, 0.3 µL of 250 mM ATP, 0.87 µL of 100 µM phalloidin, 1.91 µL of 5-488-tubulin, 0.3 µL of 100 mM GTP, and 0.75 µL of 200 µM Taxol, to a total volume of 23 µL.
- Prepare sample chambers for confocal imaging experiments as described below.
NOTE: Complete steps 2.1.4 and 2.2.2 during waiting periods.- Place two silanized slides side-by-side on a hot plate (turned off), lay two strips of thermoplastic sealing film across the slides ~3 mm apart, and place two silanized coverslips over the thermoplastic sealing film to form a sample chamber.
- Turn the hot plate on low setting until coverslips firmly bind to slides with melted thermoplastic sealing film (~1-2 min). Press down with even pressure to ensure bonding while maintaining ~100 µm spacing between the two surfaces.
- Remove chambers and turn off hot plate. Label chambers with (+) and (-). The (+) chamber will be for the active sample (with myosin) and the (-) chamber will be the control (no myosin). Ensure each chamber can accommodate ≤10 µL of fluid.
- Prepare samples to image as described below.
NOTE: It is important to complete this step immediately after steps 2.1 and 2.2 are completed.- Carefully remove the myosin-actin sample from the ultracentrifuge (step 2.1.4) and immediately pipet up the top 7.5 µL of the supernatant and transfer to a new microcentrifuge tube.
- Remove the actin-microtubule sample from the heat block and gently mix in 1.5 µL of 10x D-Glucose, 1.5 µL of 10x GOC, and 1.5 µL of 1 mM blebbistatin. Divide the solution into two 13.7 µL aliquots and label as (+) and (-).
- Mix in 1.28 µL of the supernatant from step 2.4.1 to (+) aliquot. Mix in 1.28 µL of DI to the (-) aliquot. Slowly flow each solution into the corresponding chamber (step 2.3) via capillary action. Be careful not to introduce air bubbles into the channel.
- Seal the two open ends of each channel with fast-drying epoxy or UV glue. Ensure the adhesive is completely dry before placing on the microscope. Image immediately as described in step 3.
NOTE: UV glue is advantageous because it cures nearly instantly upon UV exposure. However, because blebbistatin is UV sensitive, it is important to only locally illuminate the glue (at the edges of the sample chamber) using a small UV wand to avoid deactivating the blebbistatin.
- Optional: vary the protein concentrations to tune the dynamics and structure of the composites.
NOTE: The following steps are suggested alterations to the steps above to vary the concentrations of actin, microtubules, and myosin if desired.- Follow the steps described above except for the following modifications in steps 2.2.1 and 2.4.3.
- To vary the concentrations of actin and microtubules, thereby adjusting c and ΦA, increase or decrease the volume of actin, R-actin, and 5-488-tubulin used in step 2.2.1, as desired26. When varying the actin concentration, adjust R-actin and phalloidin molar concentrations proportionally to maintain the same molar ratios with actin. Adjust the volume of PEM such that the final volume of the mixture remains 23 µL. All other component volumes and concentrations remain the same.
- To vary the myosin concentration, adjust the volume of myosin added to the (+) aliquot in step 2.4.3 as desired. Adjust the DI volume added to the (-) aliquot accordingly. Adjust the PEM volume in Step 2.2.1 to account for the increase or decrease in myosin (+) and DI (-) volume, ensuring the final volume of each sample ((+) and (-)) is 14.98 μL.
3. Imaging and characterization of active composites using confocal microscopy
- To image actomyosin-microtubule composites prepared in step 2, use a laser scanning confocal microscope (LSCM), or similar microscope, with a 60x 1.4 NA oil-immersion objective. To simultaneously visualize actin filaments and microtubules in separate fluorescence channels, use a 561 nm laser with 565/591 nm excitation/emission filters and a 488 nm laser with 488/525 nm excitation/emission filters.
- Place the sample chamber on the microscope such that the control channel is positioned directly over the objective. Make sure there is an oil interface between the objective and the coverslip.
- Use the stage controls to bring the control composite into focus, then find both surfaces of the sample chamber. Move the z position to the center of the sample chamber. Check for the presence of clear filamentous networks as shown in Figure 2.
- Still visualizing the control chamber, adjust the intensity of each laser to allow for simultaneous visualization of actin filaments and microtubules. Maintain the lowest laser intensity possible to prevent photobleaching (more prevalent in the actin channel) and bleed through (typically from the microtubules into the actin channel).
- To characterize the inactive control sample, collect three time-series (videos) of 256 x 256 square-pixel (213 µm x 213 µm) images at 2.65 fps for a total of ≥1000 frames. Collect each time-series in a different region of the sample chamber separated by ≥500 µm. Ensure that there is minimal detectable motion and no flow or restructuring.
- Shutter off the 488 nm laser and use the stage controls to move to the (+) chamber.
- Using the 568 nm laser, visualize the microtubules in the (+) channel to ensure proper network formation (Figure 2) and identify the axial center of the sample chamber (which may be different to the center z-position of the control chamber).
- Turn on the 488 nm laser and repeat step 3.5 above with the following modifications. Collect time-series for up to 45 min, stopping acquisition when the sample either moves out of the field of view, ruptures, or photobleaches. Record 5-10 time-series and keep track of the time at which each time-series begins relative to the start of the first time-series.
- Analyze data using DDM, SIA, and PIV as described in Figure 3, Figure 4, Figure 5, and previously17,48,50,51.
NOTE: The 488 nm laser locally activates myosin ATPase activity by de-activating blebbistatin, so it should only be turned on at the start of data acquisition such that t = 0 is at the start of the time-series. These acquisition parameters are optimized for differential dynamic microscopy (DDM) analysis as done previously26.
4. Preparation of active actin-microtubule composites driven by kinesin motors
NOTE: The following steps create actin-microtubule composites that are driven out-of-equilibrium by kinesin motors or a combination of kinesin and myosin50.
- Prepare kinesin and myosin motors as described below.
- If incorporating myosin, follow step 2.1.
- To form kinesin motor clusters that bind and exert forces between pairs of microtubules, use a micropipette and sterile pipet tips to add the following to a sterile 1.5 mL microcentrifuge tube:1.16 µL PEM, 2.74 µL 8.87 µM kinesin dimers, 7.29 µL 83.3 µM NeutrAvidin, 0.81 µL 2mM DTT . Mix gently by pipetting the solution up and down and incubate protected from light (use a black microcentrifuge tube or wrap in foil) for 30 min at 4 °C.
NOTE: The molar ratio of kinesin dimers to NA is 1:25.
- Follow step 2.3 to prepare sample chambers and make three chambers instead of two. Carry out this step during kinesin incubation (step 4.1.2) and myosin ultracentrifugation (step 4.1.1).
- Prepare co-entangled composite network of actin filaments and microtubules.
- Set heat block to 37 °C. Use a micropipette and sterile pipet tips to add the following to a sterile 1.5 mL microcentrifuge tube: 3.21 µL PEM, 4.5 µL of 1% Tween20, 2.18 µL of 47.6 µM actin, 3.46 µL of 5-R-tubulin, 4.5 µL of 100 mM ATP, 4.5 µL of 10 mM GTP, 1.13 µL of 200 µM Taxol, and 1.57 µL of 20 µM 488-phalloidin. Ensure the total volume is 25 µL.
- Gently pipet the solution up and down to mix and place on the 37 °C heat block protected from light for 1 h. Remove the tube from the heat block and use a micropipette to gently mix in 0.84 µL of 100 μM phalloidin. Incubate for 5-10 min at room temperature, protected from light.
NOTE: Adding phalloidin at this step, rather than in step 4.3.1, improves the fluorescence labeling of actin filaments, as 488-phalloidin does not have to compete with unlabeled phalloidin for actin binding sites.
- Prepare active composites for confocal imaging.
- Add 1.13 µL of 200 μM blebbistatin, 1.35 µL of 10x Glu, and 1.35 µL of 10x GOC to the solution from step 4.3.2 and mix gently by pipetting up and down. Divide the solution into three 10 µL aliquots and label as (K), (K+M), and (-).
- Mix in 2.54 µL of myosin from step 2.1.4 to the (K+M) aliquot. Mix in 2.54 µL PEM to (K) and (-) aliquots.
- Use a micropipette and sterile pipet tips to add 2.5 µL of kinesin clusters from step 4.1.2 to (K) and (K+M) aliquots. Pipet up and down to mix. Mix in 2.5 µL PEM to (-) using the same technique.
NOTE: The concentrations of actin and tubulin listed are for a composite with 2.32 µM actin and 3.48 µM tubulin. Total protein concentration is c = cA + cT = 5.8 μM and molar actin fraction is cA/(cA + cT) = ΦA = 0.4. Kinesin and myosin concentrations are 0.35 µM and 0.47 µM, respectively. See step 2.5 for general guidelines to adjust cA, cT, c, and ΦA. - Using a micropipette, slowly flow each solution into the corresponding channel of the prepared sample chambers (step 4.2) via capillary action. Push down very slowly and gently on the pipet so as to not introduce air bubbles into the channel.
- Seal the two open ends of each channel with fast-drying epoxy or UV-curable glue. Ensure the adhesive is completely dry before placing on the microscope.
NOTE: It is important that this step is done quickly to minimize the time that the kinesin is acting without being monitored. For this reason, epoxy that cures in 1 min (rather than 5 or 10 min) is recommended. UV-curable glue is advantageous in this regard because it cures nearly instantly upon UV exposure.
- Image prepared samples immediately, following step 3, except for the following important modifications. Because kinesin is not controlled by light-activation, it starts to work immediately after step 4.4.3, so mark this time as t = 0. To image the composite as close to the initial inactive state (t = 0) as possible, image the (K) and (K+M) channels first and note the time elapsed between step 4.4.3 and the beginning of data acquisition (step 3.8). In practice, this elapsed time is ~5 min.
5. Incorporating passive crosslinkers into active composites
NOTE: These steps describe how to use biotinylated actin and tubulin subunits and NeutrAvidin (NA) to passively crosslink actin to actin (A-A) or microtubules to microtubules (M-M) in the active composites described in step 4.
- Prepare A-A or M-M crosslinker complexes with biotinylated proteins (biotin-actin or biotin-tubulin), NA, and biotin at a ratio of 2:2:1 biotin-actin/tubulin:biotin:NA. Start this process before Step 4 .
- For A-A crosslinkers, use a micropipette and sterile pipet tips to add 2 µL of 11.6 µM biotin-actin, 1.39 µL of 8.33 µM NA, 2.27 µL of 1.02 µM biotin, and 4.34 µL of PEM to a microcentrifuge tube. Mix gently by pipetting up and down.
- For M-M crosslinkers, use a micropipette and sterile pipet tips to add 1.86 µL of 4.55 µM biotin-tubulin, 1.11 µL of 8.33 µM NA, 1.82 µL of 1.02 µM biotin, and 5.21 µL of PEM to a microcentrifuge tube. Mix gently by pipetting up and down.
- Wrap the tube(s) from step 5.1.1 and/or 5.1.2 in thermoplastic sealing film to create a water-tight seal. Place in a flotation raft in a temperature-controlled sonicator bath set to 4 °C.
- Sonicate for 90 min at 4 °C. In practice it is best to put the sonicator in a cold room and add ice packs to the sonication bath to maintain low temperature.
- To incorporate crosslinker complexes into samples for imaging, follow step 4.3, modifying step 4.3.1 as described below for A-A crosslinking (step 5.2.1) or M-M crosslinking (step 5.2.2).
- For A-A crosslinking, combine the following in a microcentrifuge tube: 1.94 µL of PEM, 4.50 µL of 1% Tween20, 2.18 µL of 47.6 µM actin, 3.46 µL of 45.5 µM 5-R-tubulin, 1.13 µL of A-A crosslinkers (step 5.1.1), 4.50 µL of 100 mM ATP, 4.50 µL of 10 mM GTP, 1.13 µL of 200 µM Taxol, and 1.57 µL of 20 µM 488-phalloidin. Ensure the total volume is 25 µL.
- For M-M crosslinking, combine the following in a microcentrifuge tube: 1.97 µL of PEM, 4.50 µL of 1% Tween20, 2.18 µL of 47.6 µM actin, 3.76 µL of 45.5 µM 5-R-tubulin, 1.13 µL of 1:4 dilution of M-M crosslinkers (step 5.1.2), 4.50 µL of 100 mM ATP, 4.50 µL of 10 mM GTP, 1.13 µL of 200 µM Taxol, and 1.57 µL of 20 µM 488-phalloidin. Ensure the total volume is 25 µL.
- Follow steps 4.3.2-4.5 with the specific concentrations for a crosslinker:actin molar ratio of RA = 0.02 and crosslinker:tubulin molar ratio of RT = 0.005. These RA and RT values result in similar lengths between crosslinkers along actin filaments and microtubules (dA
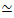 60 nm and dMT 67 nm), estimated using dA = Imonomer/2RA, where Imonomer is the length of an actin monomer, and dMT = Iring/26RT, where Iring is the length of a ring of 13 tubulins15,17.
60 nm and dMT 67 nm), estimated using dA = Imonomer/2RA, where Imonomer is the length of an actin monomer, and dMT = Iring/26RT, where Iring is the length of a ring of 13 tubulins15,17.
To determine successful preparation of active composites (Figure 1), and to characterize their dynamics and structure, a laser scanning fluorescence microscope with at least two fluorescence channels is used to visualize the actin filaments and microtubules simultaneously (Figure 2 and Figure 6). All actin filaments and microtubules in the composites are sparsely labeled, rather than doping in tracer bright filaments, as is often done in in vitro studies. This method ensures that the measured dynamics and structure are representative of the composite itself rather than the tracers which are formed under different conditions than the composites. For this reason, individual actin filaments and microtubules cannot typically be resolved, rather images portray mesoscale network structure (Figure 2 and Figure 6).
This labeling approach was optimized for spatial image autocorrelation (SIA) and differential dynamic microscopy (DDM) analyses that examine the dynamics and structure in reciprocal Fourier space (Figure 4, Figure 5, and Figure 8)52,53,54,55. Particle-image velocimetry (PIV) can also be used to depict and characterize dynamics and flow fields (Figure 3 and Figure 7), but it requires pixel-binning (lower spatial resolution) and larger lag-time increments (lower temporal resolution) than SIA and DDM to eliminate erroneous vectors that arise from noise in the dense, low-signal images. Nevertheless, PIV is recommended for qualitative examination of flow fields and corroboration of DDM results (Figure 4 and Figure 8)26,50.
Sample characterization of the described networks using these analyses (i.e., DDM, SIA, PIV) is provided to aid researchers in adopting similar analyses to benchmark and characterize their samples. However, detailed descriptions of these techniques are outside the scope of this work. For detailed descriptions of how to perform DDM on these and other similar systems, including user-friendly Python code, refer to previous works17,26,49,50 and the references there within. For details regarding how to perform SIA and PIV on the systems described here, the reader is directed to previous works17,50.
Several controls, described below, should be done to ensure that the composites are functioning as expected. A composite without myosin or kinesin should appear essentially static with minimal thermal fluctuations or drift. Actin filaments and microtubules should appear co-entangled and homogeneously distributed, with minimal bundling, aggregation, or phase separation of actin and microtubules throughout a field of view of ~200 µm x 200 µm (Figure 2, far left)17. One should expect a similar result for composites that contain myosin but are not exposed to 488 nm light (to deactivate the blebbistatin).
Upon incorporation of myosin and exposure to 488 nm light, the composites undergo contraction that is largely isotropic and similar for actin and microtubules, as seen in microscope images taken before and after myosin activity (Figure 2), as well as corresponding PIV flow fields for varying times during activity (Figure 3). To determine if the motion is ballistic, diffusive, subdiffusive, etc., the characteristic decorrelation time τ(q) determined from DDM is evaluated as a function of wave vector (i.e., reciprocal space). See as described in detail previously17,26,49. Figure 4 also demonstrates how to use DDM to characterize these composites. Power-law scaling τ(q)~1/vqβ, with β = 1, indicates ballistic motion with speed v. For reference, β = 2 represents diffusive dynamics with v being the diffusion coefficient. All active composites exhibit ballistic scaling (Figure 4A) with speeds that are tuned by the concentrations of actin and myosin (Figure 4B), and that can vary in time during activity, either accelerating or decelerating (Figure 4C,D).
Network restructuring and clustering, visible in Figure 2 and more evident for higher actin and myosin concentrations, can be characterized using SIA, as depicted in Figure 5, and described previously17,48,50. Briefly, a correlation length ξ, which is a measure of the characteristic size of features in an image, can be determined by fitting each spatial intensity autocorrelation curve g(r) to an exponential function of distance r between pixels. Larger g(r) peaks that persist for longer distances indicate larger structural features (i.e., bundling, clustering of the individual filaments). As shown in Figure 5, for higher actin fractions and myosin concentrations, significant restructuring and aggregation is reflected in the increase in ξ over time.
The viscoelastic properties and nonlinear mechanical response of the active composites can also be measured using optical tweezers microrheology (OTM). However, protocols and representative results for these experiments are outside the scope of this work. Interested readers are referred to previous works48,56 which thoroughly describe how to perform OTM measurements and the expected results.
Using the same program of experimental and analysis tools described above, the following section describes how the dynamics and structure change when kinesin motors and biotin-NA crosslinkers are incorporated into the composites (Figure 6, Figure 7, and Figure 8). Figure 6 shows representative confocal images of composites driven by either kinesin-only (K) or kinesin and myosin (K+M), with and without passive crosslinking (XL) of actin filaments or microtubules.
Incorporating kinesin into composites initially results in similar dynamics and restructuring as myosin-driven composites as seen in the top row of Figure 7 (Class 1). However, the dynamics typically transition to large-scale anisotropic flow (Figure 7 middle row, Class 2), acceleration, and deceleration (Figure 7 bottom row, Class 3). These characteristics couple with mesoscale clustering and aggregation after 5-30 min (Figure 6 and Figure 8B). PIV-generated flow fields and temporal color maps shown in Figure 7 depict examples of isotropic restructuring (Class 1, top panel), directed flow (Class 2, middle panels), and bi-directional acceleration (Class 3, bottom panels).
Speeds of actin and microtubules at varying time points during activity, determined via fits to τ(q) curves, illustrate acceleration followed by deceleration (Figure 8), which depends on crosslinking. As also shown in Figure 8, when both motor proteins are incorporated, the dynamics are actually slower than kinesin-only composites, and there is delayed onset of mesoscale flow. Myosin also supports more homogeneous interpenetration of actin and microtubule networks throughout the duration of activity, as well as less aggregation and restructuring. These effects can be seen in the images in Figure 6 and are quantified by the time-varying correlation lengths computed via SIA, which are generally smaller in the presence of myosin (Figure 8B).
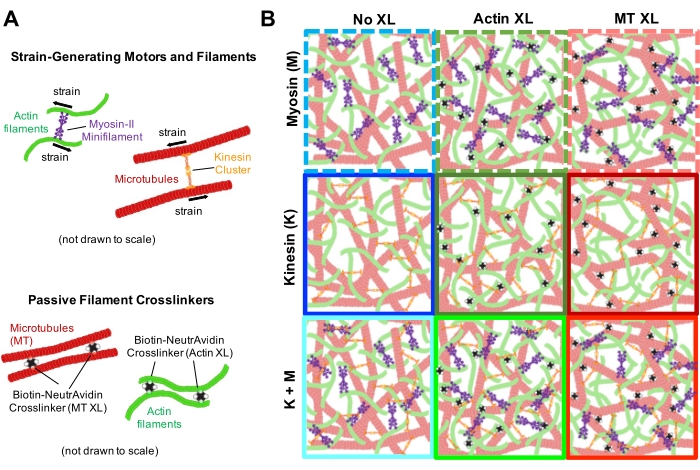
Figure 1. Design and characterization of active actin-microtubule composites with multiple force-generating motors and passive crosslinkers. (A) Actin monomers and tubulin dimers are co-polymerized at molar concentrations cA and cT of 0.73-11.6 μM and molar fractions of actin ΦA = cA / (cA + cT) = 0, 0.25, 0.5, 0.75, and 1, to form co-entangled networks of actin filaments (green) and microtubules (red). Passive crosslinking is achieved using NA to link biotinylated actin filaments (Actin XL) or microtubules (MT XL) at crosslinker:protein molar ratios of RA = 0.01-0.08 and RMT = 0.001-0.01 for actin and microtubules, respectively. Myosin-II mini-filaments (purple) and kinesin clusters (orange), at concentrations of cM = 0.12 – 0.48 μM and cK = 0.2 – 0.7 μM, push and pull on the filaments to drive the composites out of steady-state. (B) Schematic of formulation space. Myosin II mini-filaments (M), kinesin clusters (K), or both motors (K+M) are incorporated into composites with no passive crosslinkers (No XL), actin-actin crosslinks (Actin XL), and microtubule-microtubule crosslinks (MT XL). All cartoons are not drawn to scale. Please click here to view a larger version of this figure.
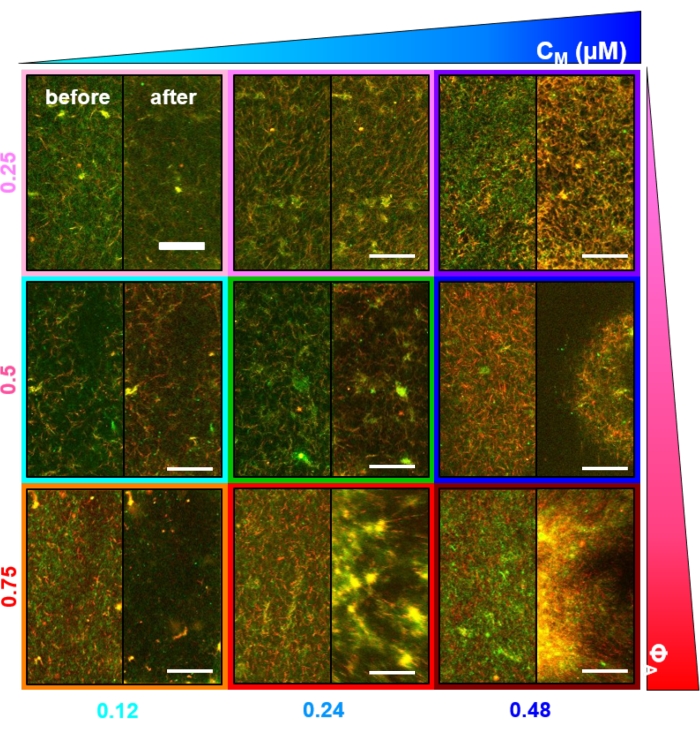
Figure 2. Two-color confocal imaging of myosin-driven cytoskeleton composites with varying myosin concentrations cM and molar actin fractions ΦA. (A) 256 x 128 square-pixel (212 x 106 μm2) two-color confocal microscopy images show how composites of actin filaments (green) and microtubules (red) are rearranged via myosin motor activity. No kinesin motors or passive crosslinkers are present. In each panel, images taken at the beginning (left, before) and end (right, after) of the 45 min myosin activation (via illumination with 488 nm light to deactivate blebbistatin) are shown. Panels are ordered by increasing molar concentration of myosin (cM), going from left to right, and increasing molar fraction of actin (ΦA), going from top to bottom. The colors outlining each panel match the color coding used in Figure 4 and Figure 5. Scale bars are 50 μM. To best capture dynamics and structure for analysis, we use frame rates of 1-5 fps, ROIs with 50-250 µm sides, and time-series durations of 5-45 min, depending on the rate of contraction and rearrangement. Panels in which the before and after images look similar indicate minimal restructuring, as seen in the pink, magenta, and cyan panels. Small-scale clustering, evidenced by increased heterogeneity and the presence of bright punctate features, can be seen in the orange, green, and red panels. Large-scale contraction, seen as a uniformly shrinking network, is evident in the blue and purple panels. This figure has been modified from reference17. Please click here to view a larger version of this figure.
Figure 3. Particle image velocimetry (PIV) shows that actomyosin activity triggers coordinated contractile dynamics of actin and microtubules in co-entangled composites. PIV flow fields for actin (top row) and microtubules (bottom row) in a myosin-driven composite with (ΦA, cM) = (0.5, 0.24) at increasing times during a 6 min time-series. Flow fields were generated using the Fiji/ImageJ PIV plugin with a lag-time of 20 s and 2 pixel x 2 pixel binning. Both actin and microtubules show consistent motion directed towards the center region of the field-of-view throughout the movie duration. Scale bars in all images are 50 μm. Different arrow colors correspond to different speeds as indicated in the color scale to the right of vector fields. This figure has been modified from reference26. Please click here to view a larger version of this figure.
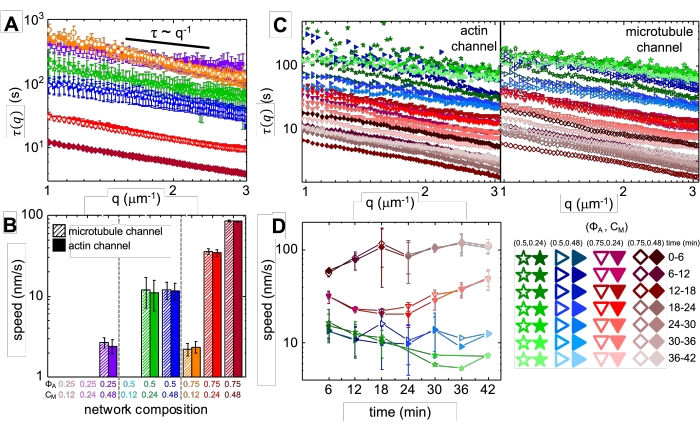
Figure 4. Time-resolved differential dynamic microscopy (DDM) measures the rate and type of motion of actin and microtubules in active composites. (A) DDM is performed on microtubule (top, open symbols) and actin (bottom, filled symbols) channels of time-series to determine characteristic decay times τ vs wavenumber q for both actin (filled symbols) and microtubules (open symbols) as described previously17,26. All curves follow τ ~ q-1 scaling, indicating ballistic motion, with speeds v that are determined via fits to τ(q) = (vq)-1. Faster speeds correspond to smaller τ(q) values for any given q. Symbol colors and shapes correspond to (ΦA, cM) combinations shown in B. (B) Contraction speeds v are determined via fits to τ(q) curves shown in A, which are averaged over all lag-times for the duration of each 45 min time-series. (C) Time-resolved DDM (trDDM) quantifies how the dynamics vary over time by evaluating τ(q) for actin (filled symbols, left) and microtubules (open symbols, right) for consecutive 6 min intervals (denoted by different shades of the same color) during the 45 min activation time. trDDM is performed for each (ΦA, cM) combination (denoted by different symbols and colors) as described in the legend in lower right. τ(q) curves shown in C follow similar scaling and trends as those in A but also show time-dependence for certain (ΦA, cM) compositions, most notably for ΦA = 0.75. (D) Contraction speeds for actin filaments (closed symbols) and microtubules (open symbols) are determined from fits to corresponding τ(q) curves. Error bars in all plots represent the standard error of values across three to five replicates. This figure has been modified from reference17. Please click here to view a larger version of this figure.
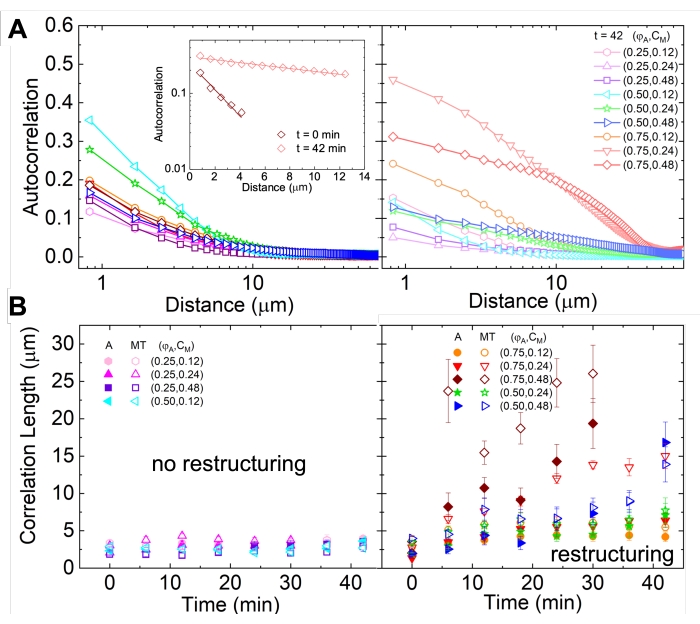
Figure 5. Spatial image autocorrelation (SIA) analysis quantifies the motor-driven restructuring of active cytoskeletal composites. (A) Autocorrelation g(r) for the microtubules at the beginning (left, t = 0 min, dark shades) and end (right, t = 42 min, light shades) of the experiment for (ΦA, cM) formulations listed in the legend. Inset: example fits of data to 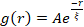 at the initial and final times for (ΦA, cM) = (0.75, 0.12). (B) Average correlation lengths ξ for actin (closed symbols) and microtubules (open symbols) for each (ΦA, cM) determined via exponential fits of each g(r) curve, as shown in the inset in A. Data is divided into those that exhibit minimal (left) versus substantial (right) restructuring. Error bars in A and B represent the standard error across three to five replicates. This figure has been modified from reference17. Please click here to view a larger version of this figure.
at the initial and final times for (ΦA, cM) = (0.75, 0.12). (B) Average correlation lengths ξ for actin (closed symbols) and microtubules (open symbols) for each (ΦA, cM) determined via exponential fits of each g(r) curve, as shown in the inset in A. Data is divided into those that exhibit minimal (left) versus substantial (right) restructuring. Error bars in A and B represent the standard error across three to five replicates. This figure has been modified from reference17. Please click here to view a larger version of this figure.
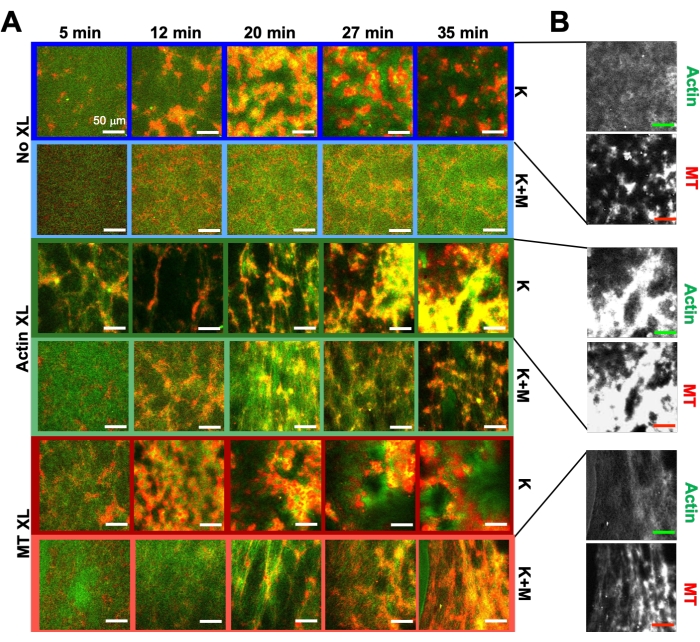
Figure 6. Incorporating kinesin motors and passive crosslinkers into active composites to increase programmability and expand the phase space of dynamics and structure. (A) Two-color confocal images of actin (green) and microtubules (red) in active composites show complex formulation-dependent restructuring over time (listed in min). The five images in each row correspond to five frames of a 2000 frame time-series acquired for a composite driven by kinesin (K, rows 1, 3, 5) or kinesin and myosin (K+M, rows 2, 4, 6), and including either no passive crosslinkers (No XL, rows 1, 2), actin-actin crosslinks (Actin XL, rows 3, 4), or microtubule-microtubule crosslinks (MT XL, rows 5, 6). Scale bars are all 50 µm. Outline colors match color scheme in Figure 8. (B) Separate actin and microtubule fluorescence channels for the kinesin-only composites show varied structures with both actin-MT co-localization and micro-phase separation. The images shown are for composites with cA = 2.32 μM, cT = 3.48 μM, cK = 0.35 μM, cM = 0.47 μM (rows 2, 4, 6), RA = 0.02 (rows 3, 4), and RMT = 0.005 (rows 5, 6). All composites begin with uniformly distributed interpenetrating networks of actin and microtubules (column 1). Kinesin-driven composites without crosslinkers (row 1) form loosely connected amorphous clusters that are MT-rich. Actin co-localizes in the centers of these aggregates initially but then is squeezed out of the MT-rich regions that continue to contract and disconnect from one another. Actin-actin crosslinking (row 3) hinders this microscale actin-MT separation, and instead MT-rich aggregates are connected via long strands of actin. Actin crosslinking also enables slow uptake of actin into the MT-rich regions, such that the composite becomes a connected network of co-localized actin and MT clusters. Microtubule crosslinking (row 5) leads to amorphous clustering of MTs that coalesce over time, resulting in larger scale phase separation of actin and MTs. Adding myosin (rows 2, 4, 6) reduces kinesin-driven de-mixing and restructuring. Without crosslinkers (row 2), composites show little rearrangement over the course of hours. Crosslinking increases restructuring and co-localization of actin and microtubules (rows 4, 6). Specifically, when microtubules are crosslinked (row 6), there is significant interpenetration and reorganization into web-like networks of fibers. This figure has been modified from reference50. Please click here to view a larger version of this figure.
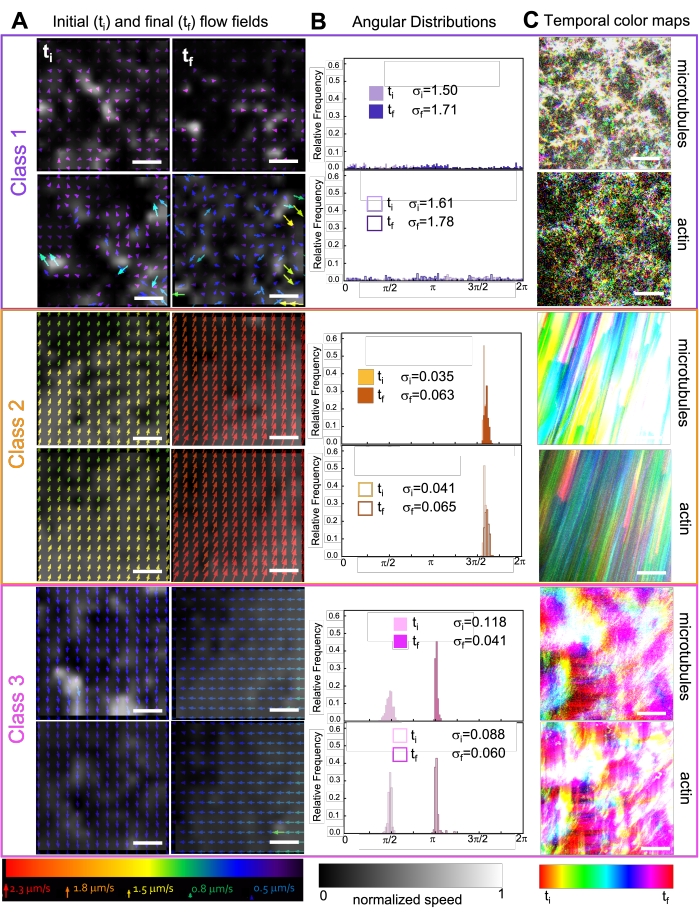
Figure 7. PIV shows that active composites exhibit three classes of spatiotemporally distinct flow fields. (A) PIV flow fields for the first (ti) and last (tf) frames of three representative time-series, showing the different dynamical classes that composites shown in Figure 6 exhibit. PIV flow fields for microtubules (top) and actin (bottom) for class 1 (top, purple), class 2 (middle, orange), and class 3 (bottom, magenta) example videos, with arrow colors corresponding to the universal speed scale at the bottom, and the grayscale colormap showing the spatial speed distribution, normalized separately for each flow field according to the scale shown at bottom. Scale bars are all 50 μM. (B) Angular distributions of velocity vectors from A (in units of radians) with listed initial and final standard deviations σi and σf. (C) Temporal color maps for the videos analyzed in A and B show the frame-to-frame position of each pixel relative to its starting point. Class 1 maps show small-scale random motion; class 2 maps depict fast unidirectional motion with minimal spatial or temporal variation; class 3 maps exhibit features of both class 1 and 2. This figure has been modified from reference50. Please click here to view a larger version of this figure.
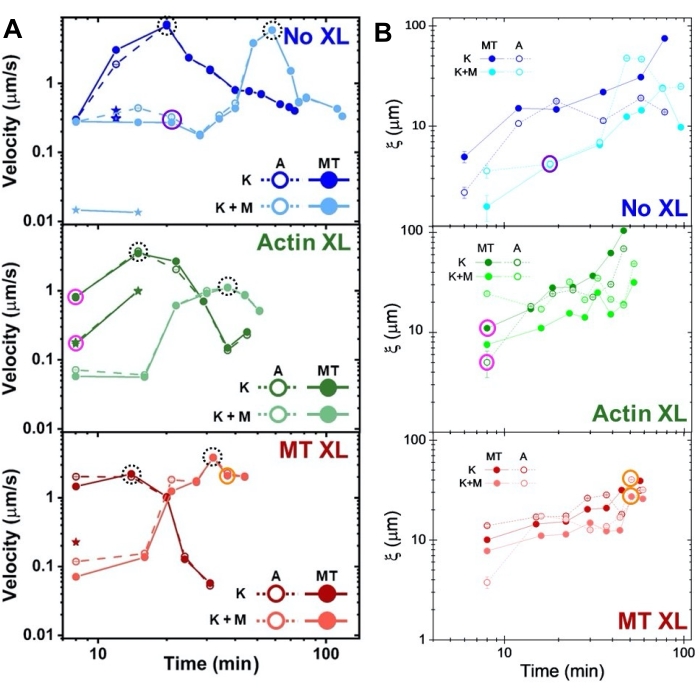
Figure 8. DDM and SIA measure the time-varying dynamics and structure of two-motor actin-microtubule composites. (A) Speeds for composites described in Figure 6 and Figure 7, measured via DDM, show acceleration and deceleration of composites, programmed by crosslinking and myosin activity. Speeds of microtubules (MT, closed circles) and actin (A, open circles) are plotted as a function of activity time in composites with no crosslinking (top, blue), actin crosslinking (middle, green), microtubule crosslinking (bottom, red), without myosin (K, darker shades), and with myosin (K+M, lighter shades). For class 3 cases, which have two speeds, the slower speed is indicated by a star. Data points enclosed by dashed black circles correspond to maximum speed vmax for each formulation. Error bars (most too small to see) are the standard error over the power-law fits of the corresponding τ(q). (B) Structural correlation lengths ξ, determined via SIA, versus activity time, for the same set of time-series evaluated in A. Each data point is an average of the correlation lengths determined for the first and last frame of the corresponding time-series. In general, ξ increases in time for both actin and microtubules in all composites systems, and composites driven solely by kinesin have greater correlation lengths than those in which myosin is also present. Data points in A and B that correspond to the three time-series analyzed in Figure 7 are circled in the corresponding class color (1 = purple, 2 = orange, 3 = magenta). This figure has been modified from reference50. Please click here to view a larger version of this figure.
