This protocol shows how to establish human skin-derived fibroblast cultures and convert them into myoblasts and then into differentiated myotubes. This type of cell line is extremely useful for the study of neuromuscular disorders and in vitro testing of potential therapies.
A schematic representation of the fibroblast conversion is shown in Figure 1. Figure 2A shows a fragment of skin and the fibroblasts emerging from it. The fibroblasts should be passed to a new dish when confluence is reached (Figure 2B). Figure 3A shows the ideal confluence of fibroblasts before changing to myoblast growth medium supplemented with doxycycline. The cells should be around 70% confluent because they still proliferate during the conversion process. If cells are above 80% confluent, the differentiation may be compromised. The conversion into myoblasts takes two to four days, and it is confirmed by observation of the morphology. The cells become elongated and parallelly oriented, as shown in Figure 3B. After the addition of the differentiation medium, the myoblasts stop dividing and start to fuse to form multinucleated myotubes (Figure 3C). When the myotubes borders look white and bright, they are about to detach (Figure 4). At this point, collect or fix the cells.
The differentiation success will vary between different cell lines/mutations. Immunostaining of muscle proteins expressed by mature myotubes confirms the myogenic potential of converted fibroblasts (Figure 5). RNA-Seq analysis comparing FM myotubes and skeletal muscle showed high-level expression of transcripts from the embryonic (MYH3) and neonatal (MYH8) myosin chain genes and good overall transcriptome-wide correlation with muscle (Figure 6). Transcripts for the giant sarcomeric proteins titin (TTN), nebulin (NEB), and obscurin (OBSCN) are also expressed by FM myotubes, indicating upregulation of these large transcripts involved in myofibrillogenesis. Thus, FM cells have a muscle-specific expression profile, demonstrating that they are a useful and reliable surrogate for muscle-derived cell lines.
To illustrate exon skipping, we used this protocol in one of the most frequent exon duplications in the DMD gene. Duplication of exon 2 leads to disruption of the DMD reading frame, thus the restoration of the reading frame following exon skipping should lead to the expression of the full-length dystrophin. However, it is also possible that skipping of exon 2 is very efficient resulting in an out-of-frame transcript. Nevertheless, in this case, skipping of both copies of exon 2 induces the utilization of an alternative internal ribosome entry site (IRES) present in exon 5, thereby producing functional N-truncated dystrophin that was identified in patients still ambulant in their 70s12. Figure 7A shows representative results of RT-PCR of FM cells with exon 2 duplication. FM cells were treated either with AON or AAV1-U7 carrying an antisense sequence to skip exon 2. In Figure 7B, an immunoblot shows the detection of the N-truncated dystrophin in FM cells treated with AAV1-U7. In vitro treatment of FM cells serves as proof of concept for exon-skipping strategies.
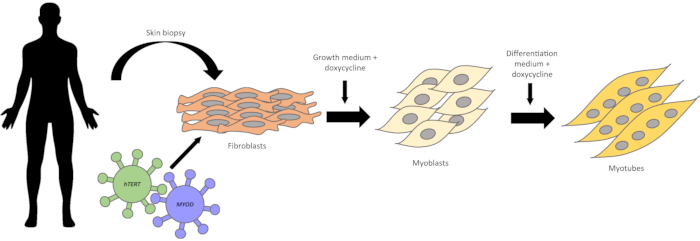
Figure 1: Schematic representation of fibroblasts conversion into myogenic cells. A skin biopsy is obtained from human subjects. Skin fragments are placed on culture dishes. Within one week, fibroblasts start to emerge. Fibroblasts are first transduced with the hTERT gene, and then with the Myod gene, using lentiviral vectors. After antibiotic selection of infected cells, the conversion into myoblasts is induced by the addition of doxycycline to the myoblast growth medium. Within two to four days, the cells become elongated and parallelly oriented. After switching to differentiation medium, the myoblast fuse with each other and form multinucleated myotubes. Please click here to view a larger version of this figure.

Figure 2: Skin biopsy fragments in culture. (A) First fibroblasts emerging from skin fragment. (B) Confluent fibroblasts emerged from the skin fragment. Scale bar: 50 µm. Please click here to view a larger version of this figure.

Figure 3: Fibroblasts transdifferentiation. (A) Representative image of 70% confluent fibroblasts. (B) Converted myoblasts have elongated morphology and are parallelly organized. (C) Myotubes were differentiated for 7 days. Scale bar: 50 µm. Please click here to view a larger version of this figure.

Figure 4: Representative image of detaching myotubes. The arrows indicate the white and bright edges of myotubes starting to detach. Scale bar: 50 µm. Please click here to view a larger version of this figure.
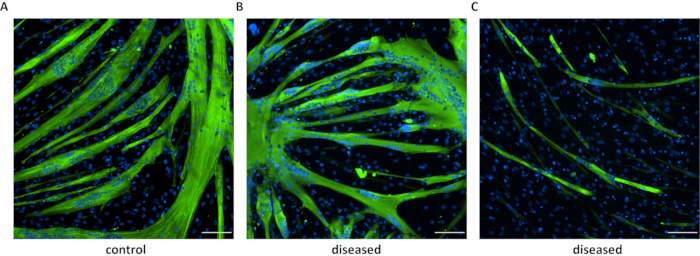
Figure 5: Immunofluorescence of differentiated myotubes. Immunostaining of myosin heavy chain in myotubes derived from a healthy (A) individual and patients with neuromuscular disorders (B and C). In B are shown cells from myotonic dystrophy type 1 (DM1) carrying 230 CTG repeats, and in C are DM1 cells with 900 CTG repeats. Scale bar: 100 µm. Please click here to view a larger version of this figure.
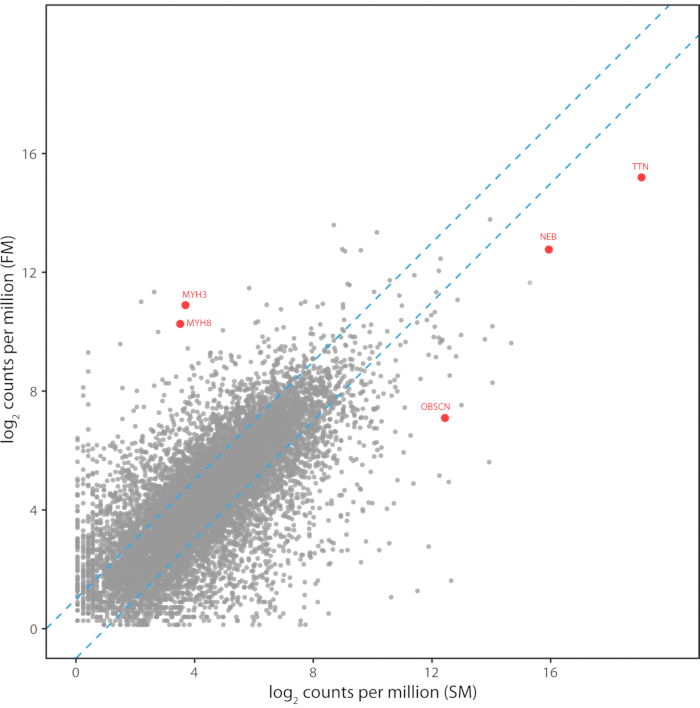
Figure 6: Transcriptome pattern of FM myotubes compared to skeletal muscle. Transcriptome pattern of FM myotubes compared to skeletal muscle. The read counts per million mapped reads for 12,134 transcripts are shown for Illumina RNA-Seq libraries prepared from FM myotubes and a human skeletal muscle biopsy. Transcript levels between the two libraries had a Pearson correlation of 0.71 and a Spearman rank correlation of 0.73. Transcripts for the developmental myosin heavy chains and the large sarcomeric proteins are highlighted in red. Please click here to view a larger version of this figure.
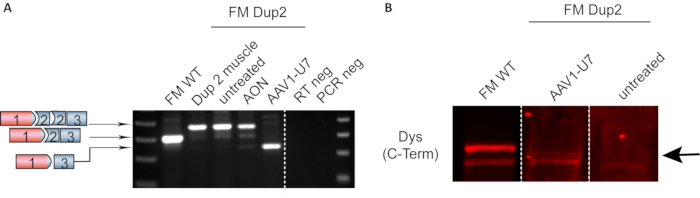
Figure 7: Representative RT-PCR and Western blot showing DMD exon skipping in FM cells. (A) Expression DMD by RT-PCR. Fibroblasts from a patient harboring a duplication of DMD exon 2 were converted into FM cells. RNA extracted from muscle biopsy was used as the control, showing that FM untreated cells express the same duplicated transcript. FM cells treated with AON have a partial skipping of exon 2 duplication, while AAV1-U7 treated cells showed a predominance of transcripts with exon 2 duplication skipped. (B) Representative immunoblot of FM cells treated with AAV1-U7. Smaller N-truncated dystrophin was detected 14 days after treatment (indicated by the arrow). Data previously published in Wein et al. Translation from a DMD exon 5 IRES results in a functional dystrophin isoform that attenuates dystrophinopathy in humans and mice. Nature Medicine. 2014. 2020 Springer Nature Limited. Please click here to view a larger version of this figure.
| Fibroblast growth medium | DMEM with 20% FBS, 1% antibiotic-antimicotic |
| Freezing medium | 10% DMSO, 90% fibroblast medium |
| Doxycycline stock solution 1000X | 8 mg of doxycycline in 1 mL ultra-pure water. Filter in 0.22 µm syringe filter. Aliquot in PCR tubes. Store at -20 °C, protected from light. |
| Myoblast medium | Skeletal muscle cell growth medium (see list above) with supplements, 8 µg/mL doxycycline. For example: 100 µL of 1000X stock solution in 100 mL. |
| Differentiation medium | Skeletal muscle cell differentiation medium with supplements (see list above), 8 µg/mL doxycycline. For example: 100 µL of 1000X stock solution in 100 mL. |
| Blocking solution for IF staining | 10% goat serum (or serum of animal in which secondary antibody was raised) in 1X PBS |
| Base buffer for protein extraction | NaCl 150 mM, Tris 50 mM, 0.05 % NP-40. Adjust pH to 7.4. Store at 4 °C. |
Table 1: Medium recipes
